INTRODUCTION
Malignant peripheral nerve sheath tumors (MPNSTs) are a group of tumors previously known as malignant schwannomas, neurofibrosarcomas, or neurogenic sarcomas [1]. MPNSTs arise from cells that comprise the nerve sheath, such as Schwann cells, perineural fibroblasts, or fibroblasts of peripheral nerves, autonomic nerves, and cranial nerves. MPNSTs occur spontaneously in 30%, whereas as the remaining 70% are associated with neurofibromatosis type 1 [1]. They comprise approximately 5% of all malignant soft-tissue tumors [2].
In this report, we describe a case of MPNST involving the frontal sinus, esthmoid sinus, and orbital cavity. To our knowledge, such MPNST has not been previously described.
CASE REPORT
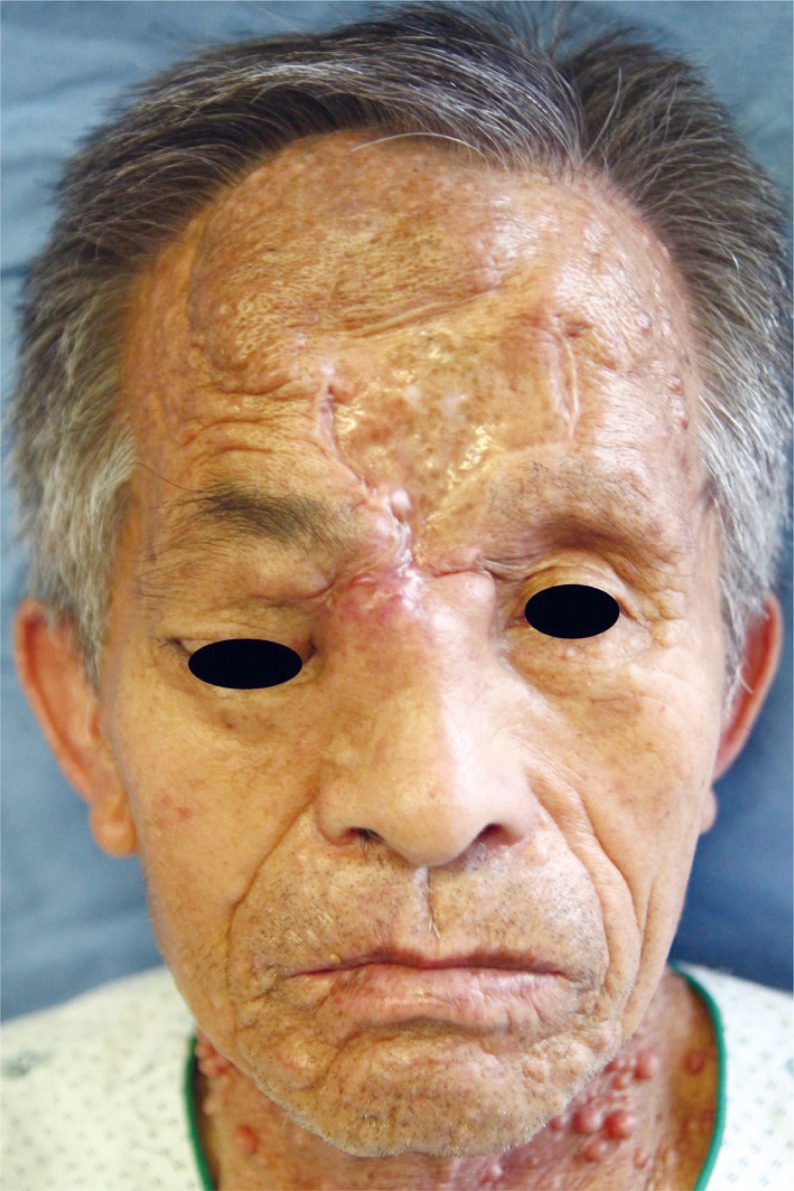
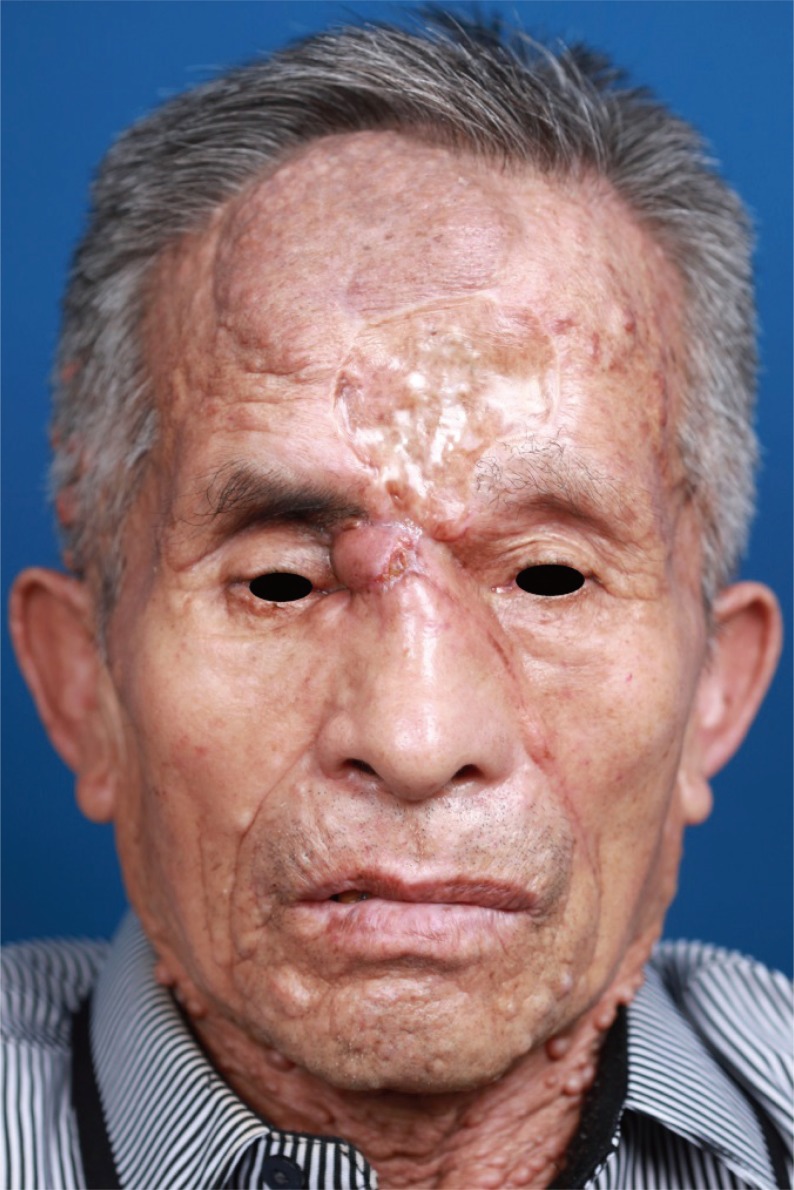
A 74-year-old man with neurofibromatosis presented with a new-onset forehead swelling and right eye deviation, which had been progressive over the past few months (Fig. 1). The patient had a history of surgical excision and full thickness skin graft for a forehead neurofibromatosis about two years ago. Subsequent to this, the tumor recurred and increased in size but was not associated with pain, headaches, epistaxis, nasal discharge or obstruction, or malodor. The patient did not report systemic symptoms such as fever or weight loss.
On examination, respective visual activity was 0.2 and 0.15 for right and left. His right eye deviation was to the inferolateral side with superior and medial gaze limitations. Diplopia was pronounced with upper gaze. The mass was about 5 × 6 cm in size, irregularly shaped, tender to palpation, and without overlying skin changes. On palpation, the mass was of mild tenderness, soft and fixed. No pulsation or bruits were observed (Fig. 1). He had café au lait spots and multiple neurofibromatosis throughout the entire body. The patient did not have any familial history significant for neurofibromatosis.
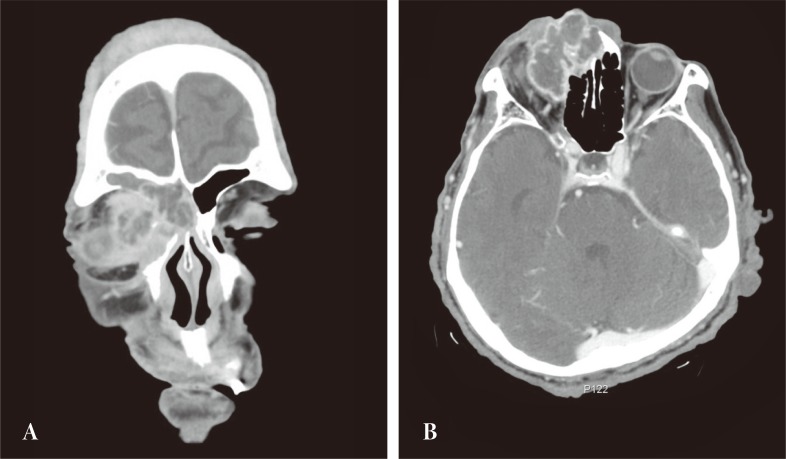
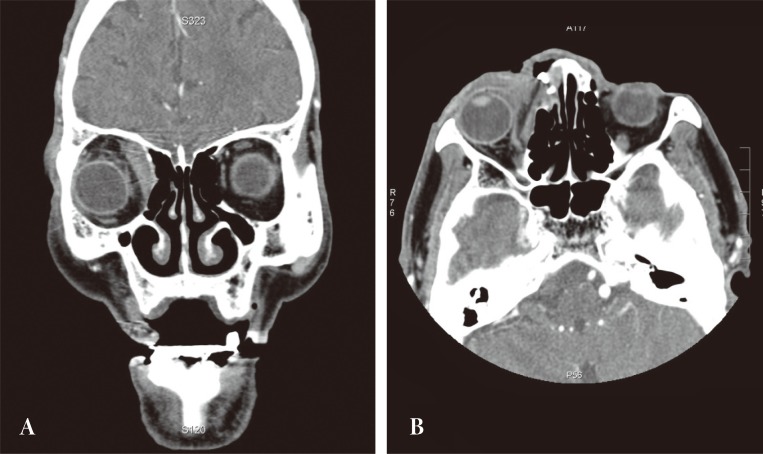
Computed tomography revealed the mass to involve the right orbital cavity as well as the frontal and ethmoid sinuses. The 4 × 4.5 cm mass was found to have a locally expansile effect, with partial erosion of the anterior, posterior frontal sinus wall, lateral ethmoid wall, and compressive right orbit. The lesion did not extend into the nasal cavity or intracranium (Fig. 2).
Under general anesthesia, the patient underwent a mass resection through the forehead scar from the previous operation. The mass was removed along with the skull base and lateral nasal wall, using suction and curettage. In the orbital cavity, the mass was dissected sharply from the surrounding tissue to preserve the ocular muscles. After mass excision, defects of the medial orbital wall and nasal bone and frontal sinus anterior wall were reconstructed with Medpor. The postoperative course was complicated by necrosis of the skin flap over the right medial canthus and nasal dorsum (1.5 × 2 cm). The resulting defect was reconstructed with a left nasolabial fold island flap under local anesthesia 2 weeks after the mass resection.
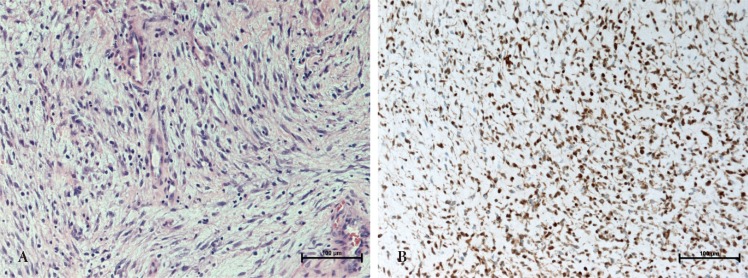
On histologic examination, the excised specimen was identified as a cellular infiltration lesion made up of pleomorphic spindle-cells. The nuclei were pleomorphic and atypical. On immunocytochemistry, the tumor cells were found to express S-100 protein and were focal positive for Ki-67. Tumor morphology and immunocytochemical phenotype were consistent with a MPNST (Fig. 3).
DISCUSSION
The incidence of malignant peripheral nerve sheath tumors is 0.001% in the general population [3,4,5,6]. MPNSTs usually occur between the third and fifth decades of life, with equal sex distribution [3,4,5,6]. MPNSTs are spontaneous in 30% of cases, and are associated with neurofibromatosis type 1 in up to 70% of cases [1]. MPNSTs arise de novo or with the development of neurofibromatosis type 1. The rate of malignant transformation is 2%-29% and accounts for about 50% of all MPNSTs [3,7]. Compared to patients with spontaneous MPNST, patients with neurofibromatosis develop these tumor at an earlier age and have poorer prognosis because there is a tendency toward larger tumors, poorer differentiation, higher rates of metastases, and multifocality in neurofibromatosis-associated tumors [4,8]. The etiology of these tumors is uncertain, but radiation exposure is known to be a general risk factor [1,9]. In this case, the patient had multiple neurofibromatoses in his whole body but without any history of radiation.
The most common symptom of MPNST is a rapidly enlarging painless mass. Other symptoms are mass effects upon the peripheral nerve, such as pain, paresthesia, and paralysis [1].
Computerized tomography and magnetic resonance imaging are useful in determining the anatomic extent of the tumor and its relationship to adjacent vital structures [10]. These imaging studies may differentiate benign tumors from malignant tumors, such as aneurysmal formations, vascular tumors, cholesteatomas, meningiomas, or fibrous dysplasia. However, they do not provide information regarding a specific histologic diagnosis [2].
On gross exam, MPNSTs are often described as firm, round masses with a light gray color. Histology reveals multiple hyperchromatic spindle cells that are arranged in the fascicles of a herring bone pattern. These spindle cells have atypical nuclei and a high mitotic rate [1,8]. MPNSTs usually demonstrate S-100 protein positivity [11]. They are reactive to vimentin, and are negative for human melanoma black 45 [12]. In our case, the tumor cells were positive for S-100 and Ki-67.
The treatment of choice in MPNSTs is wide excision. Adjuvant radiotherapy is recommended when complete removal of the tumor is not possible or when the tumor has become metastatic. Chemotherapy remains controversial in MPNST [8]. Our patient wanted symptom resolution and did not want to undergo a radical excision because of his advanced age. As a result, he did not receive radiotherapy or a metastatic work up.
Similar to other forms of sarcomas, MPNSTs infiltrate rapidly into the surrounding tissue. Lymphatic spread is rare, and tumors spread through blood vessels to the liver, lung, bone and other organs in about 39% of cases [1,5,9]. In MPNSTs, the respective survival rates for patient with and without neurofibromatosis1 are 23% and 47% at five years, with patient age, tumor location, tumor size, quality of the margins, and the extent of surgery being modifying factors [4,5,8]. In head and neck MPNST, the five year survival rate is reported to be between 15% and 47% [1].
To the best of our knowledge, this case is the first presentation of a MPNST involving the frontal sinus, ethmoid sinus, and orbital cavity. MPNSTs are extremely rare with low survival rate. Because patients with neurofibromatosis type 1 often develop malignant changes, close observation for malignant transformation is important.