INTRODUCTION
Kimura disease (KD) is a rare chronic inflammatory condition of unknown etiology that usually presents as soft tissue tumors of the head and neck, especially in the retroauricular area, scalp, and forehead of young to middle-aged Asian men. It is often accompanied by regional lymphadenopathy, elevated serum immunoglobulin E (IgE) levels, and raised serum eosinophil counts [1–4]. The histologic findings include proliferating blood vessels and eosinophilic infiltration, which suggest an inflammatory response associated with an allergic or an autoimmune reaction to an unknown stimulus [5]. Surgery enables the greatest diagnostic accuracy and has been the mainstay of treatment. However, the optimal treatment of KD remains a matter of debate, and other options besides surgery have emerged [1,5]. This article reviews the fundamental features of KD and current knowledge regarding its treatment.
HISTORY
KD was first described by Kim in 1937 [6] as an “eosinophilic hyperplastic lymphogranuloma”. A decade later, a systemic description that included the clinical entity of unusual granulation combined with hyperplastic changes of lymphatic tissues in a self-limited allergic or autoimmune process was established by Kimura [7], for whom the disease was named. Although the condition is prevalent in East Asia in an endemic form, relatively few cases have been reported in the Western literature, in African Americans and Caucasians as well as individuals of Arab ancestry [4,8–10].
HISTOPATHOLOGY
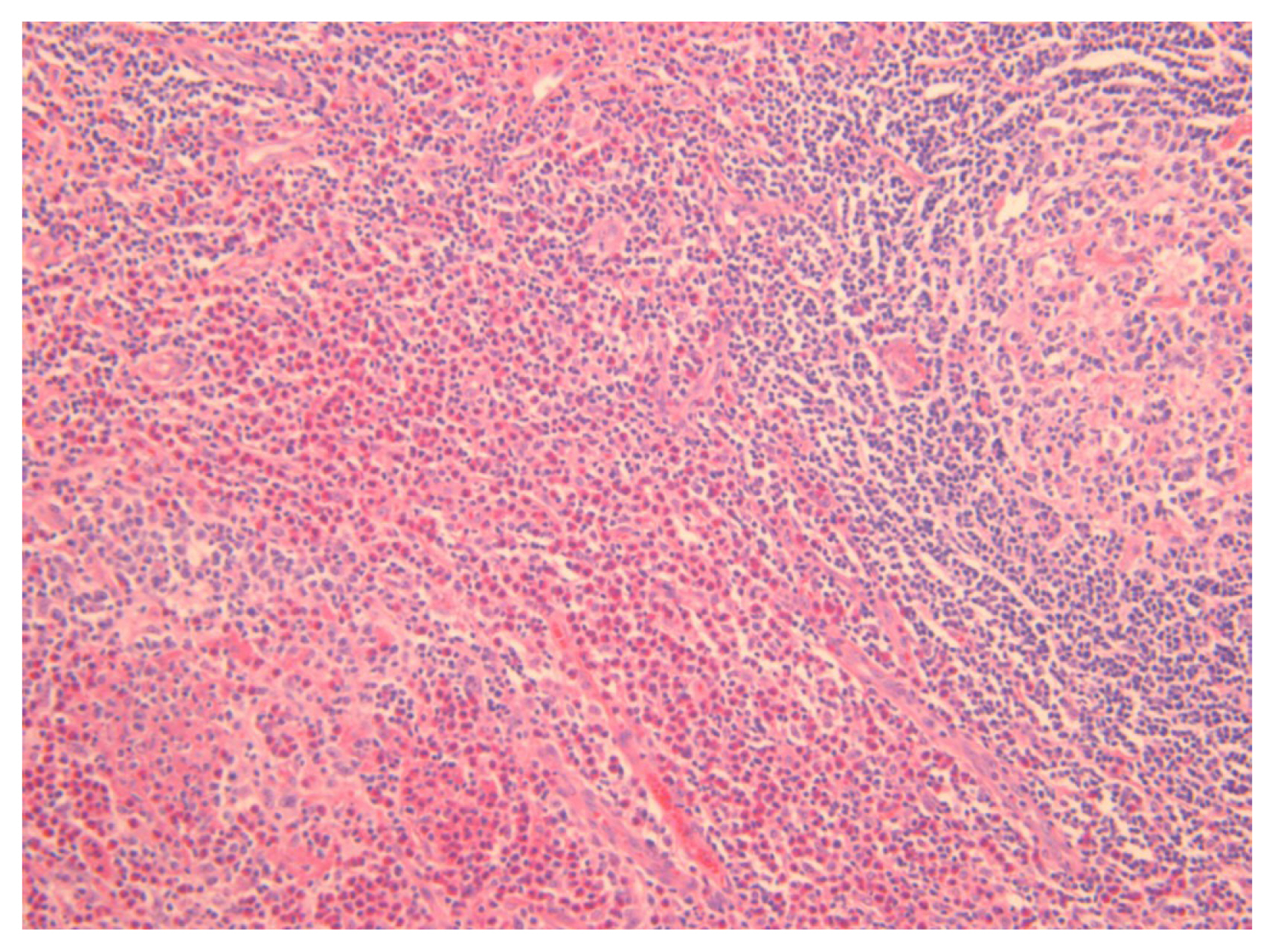
The histologic features of KD include follicular hyperplasia with reactive germinal centers; eosinophilic infiltrates involving the inner follicular areas, sinusoidal areas, perinodal soft tissues, and subcutaneous tissues; and the proliferation of postcapillary venules while preserving the nodal architecture (Fig. 1) [5,11–15]. Chen et al. [11] reviewed 21 cases of KD and observed the following histologic features; eosinophils, necrosis, proteinaceous deposits and vascularization of germinal centers, polykaryocytes, eosinophilic folliculolysis, eosinophilic micro-abscesses, stromal sclerosis, perivenular sclerosis, rare giant cells, or small eosinophilic granulomas [5,11,16]. Various degrees of sclerosis are found in KD, and immunohistochemical staining for IgE reveals a reticular pattern of germinal centers [11].
PATHOGENESIS
The etiology and pathogenesis of KD are still unknown despite numerous studies. Although increased levels of IgE, tumor necrosis factor α, interleukin (IL)-4, IL-5, IL-13 and mast cells in peripheral blood, as well as in the affected lesions with eosinophilia, have been observed in most patients, no specific antigens have been identified [17–19]. Allergic reactions, infections, neoplasms, and autoimmune reactions with an aberrant immune reaction have been suggested as possible etiologies [20–22]. However, as Lu et al. [19] reported in a single case of KD secondary to renal allograft failure involving chronic graft rejection induced by T-helper produced ILs, T-helper 2 (Th2) cytokines, overgrowth of CD4+ (cluster of differentiation 4) cells, and Th2 may play an important role in KD. Furthermore, molecular studies of the immunoglobulin heavy chain and T-cell receptor gene support this reactive process. Clonal T-cell rearrangement was also described in a case report by Chim et al. [23] and confirmed by complete sequencing of the VDJ rearrangement [5,23,24].
CLINICAL FEATURES
Most lesions in KD involve deep subcutaneous tissues of the head and neck area, presenting as one or several deeper masses. The masses are observed as non-tender and poorly circumscribed with pruritus and pigment deposition in rare cases [1,25]. Commonly involved regions include the periauricular area, epicranium, orbit, and eyelids, while other sites such as the groin, extremities, and the soft tissue of the trunk are less often affected. Rare cases have been reported involving the hard palate, the larynx, and the median nerve [9,25–27]. The majority of these masses frequently involve regional lymphadenopathy or salivary gland enlargement. However, most patients do not suffer from systemic symptoms, such as fever, night sweats, and weight loss. Coexisting renal disease from glomerulonephritis can be manifested by concomitant proteinuria and present as nephrotic syndrome with increased serum creatinine [12,28, 29]. Renal impairment might originate from immunocomplex-mediated damage or Th2 immune response disorders [28]. These renal diseases usually occur simultaneously with skin lesions [30].
DIAGNOSIS
Since there is no definite single specific diagnostic tool for KD, biopsy or excision of the involved mass or lymph node is frequently required for a pathological diagnosis. The histopathological findings of KD are similar, regardless of the anatomical site of involvement, and are characterized by a marked reactive follicular hyperplasia surrounded by a large number of eosinophils, lymphocytes and mast cells, sometimes forming micro-abscesses with vascular proliferation [16,31]. The vessels maintain thin walls of cuboidal endothelial cells, and the polykaryocytic Warthin-Finkeldey type of giant cells is a common finding. These findings are also present in lymph node biopsies, which show lymphoproliferation with prominent lymph follicle hyperplasia and germinal center enlargement while the nodal architecture is preserved [32,33].
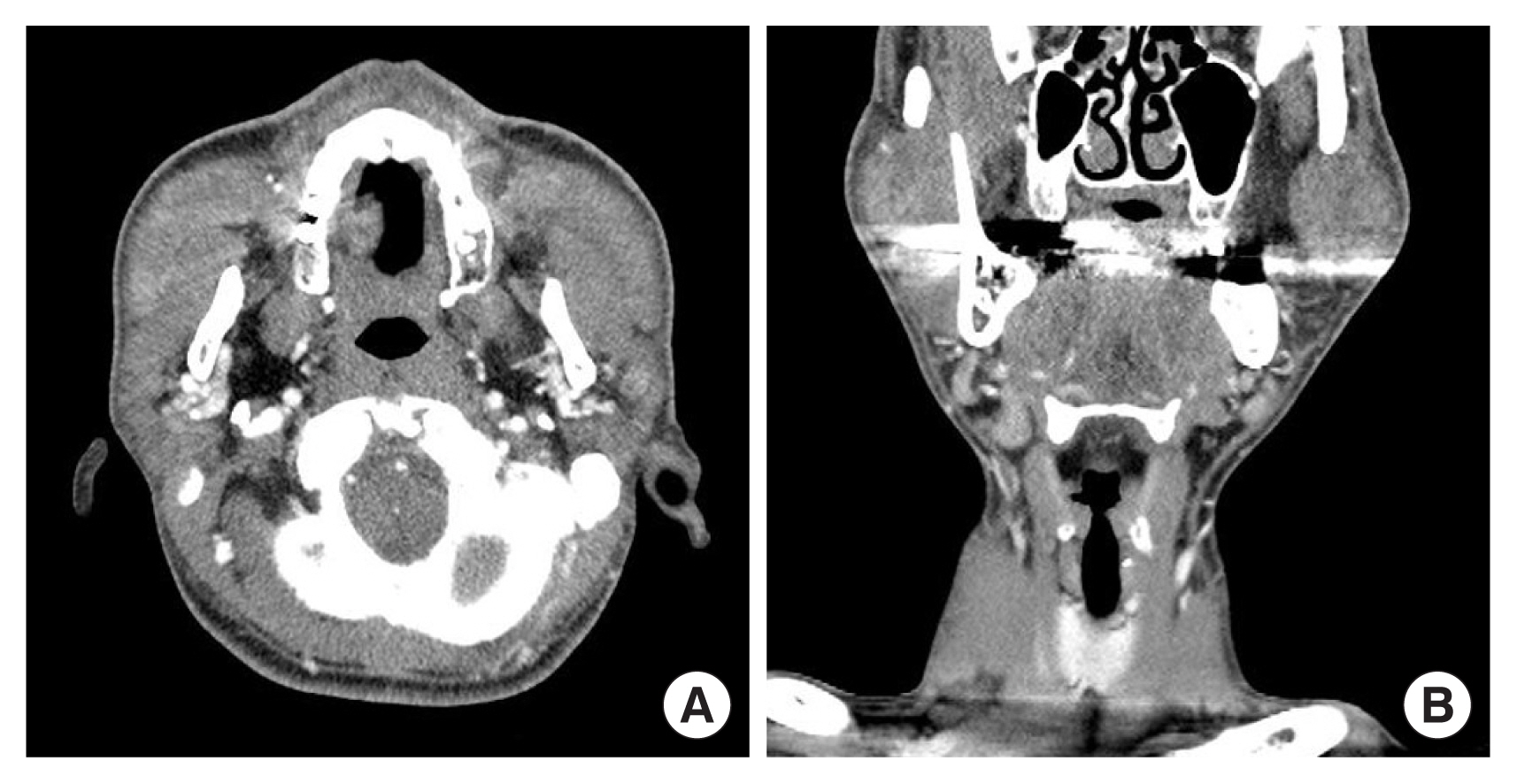
The radiologic findings of masses may aid in diagnosis of KD and differentiate them from other soft tissue tumors. On computed tomography with contrast enhancement, ill-defined enlarged subcutaneous masses with homogeneous, hyperattenuated lymph nodes and swollen salivary glands appear, reflecting the vascular nature of the lesions (Fig. 2) [29,34,35]. On magnetic resonance imaging, the lesions show heterogeneity, with both hypointense and mixed or high signal intensity on T1-weighted images and hyperintense signals on T2-weighted images. The degree of enhancement is variable and relates to the degree of fibrosis and vascular hyperplasia [35,36]. On positron emission tomography-computed tomography, KD may resemble neoplastic disorders, such as lymphoma or metastatic lymphadenopathy [37].
The differential diagnosis includes angiolymphoid hyperplasia with eosinophilia (ALHE), angioimmunoblastic T-cell lymphoma, Hodgkin lymphoma, Langerhans cell histiocytosis, Castleman disease, florid follicular hyperplasia, dermatopathic lymphadenopathy, allergic granulomatosis of Churg and Strauss, drug reaction, tumor node metastases, and parasitic lymphadenitis. KD can be confused with ALHE, which was often considered identical to KD in the past literature. However, ALHE is considered to be a type of endothelial neoplasm related to inflammatory stimulation, while KD is a chronic inflammatory disorder. ALHE often affects middle-aged Western women presenting with a well-circumscribed subcutaneous mass in the head and neck region, normally without regional lymphadenopathy, serum eosinophilia, or elevated IgE levels. Microscopically, ALHE shows the vascular endothelium forming aggregates and lobules lined by plump cuboidal or hobnail endothelial cells, which frequently involve large muscular vessels (Table 1) [7,17,38,39].
EVOLUTION OF TREATMENT
Several interventions have been reported in the previous literature; however, there is a lack of evidence on the optimal treatment for KD. Furthermore, owing to the rarity of KD, most clinical studies have been case reports or series reports; therefore, debate continues regarding the optimal treatment of KD.
Corticosteroids
Previous studies have reported the efficacy of systemic steroid therapy. Steroids are used to control local lesions, lymphadenopathy, and nephrotic syndrome. In particular, a patient with nephrotic syndrome was responsive to high-dose systemic steroids, with disappearance of proteinuria 7 months after treatment [28]. Another case report from China also revealed that the majority of patients included in the study responded well to treatment with corticosteroids alone [40]. However, Nakahara et al. [41] reported that local recurrence was frequently observed during steroid dose tapering or drug withdrawal and stated that long-term steroid use may cause digestive ulcers, osteoporosis, and acquired diabetes mellitus [41–43]. Therefore, it is reasonable to use a steroid as a second-line treatment in KD while considering the risk of recurrence and side effects.
Immunosuppressive medications
According to a few recent studies, cyclosporine A is effective for recurrent KD patients [17,41,44–48]. According to these reports, cyclosporine A was usually used for combined therapy with steroids to induce re-remission after KD relapse [49]. Cyclosporine A inhibits calcineurin, which signals IL-2 gene transcription in lymphocytes. The reduction of IL-2 inhibits T cell proliferation and suppresses T cell-mediated immune responses [17,50,51]. Moreover, Katagiri et al. [17] reported that cyclosporine A also decreased mRNA levels of IL-4, IL-5, and IL-13, resulting in the presence of fewer peripheral eosinophils and lower serum levels of IgE.
Omalizumab is an anti-IgE monoclonal antibody that binds to free IgE in the blood, which in turn, inhibits the activation of inflammatory mechanisms. As KD is suspected to be an IgE-mediated allergic disorder, a single pilot study reported evidence of omalizumab for treating KD. According to that study, the size of the masses and eosinophil counts of peripheral blood all decreased after omalizumab administration [52]. Moreover, omalizumab was also reported to be available for patients whose total serum IgE levels were below <1,500 kU/L in another case report. Therefore, omalizumab, as well as anti-IgE therapy, might have the potential to become definite treatment option for KD [53].
Imatinib, an inhibitor of protein tyrosine kinase (PTK), selectively blocks PTK and works effectively for steroid-resistant patients in hypereosinophilic syndrome. Since the tissues of KD patients are positive for expression of PTK with hypereosinophilic conditions, the authors of a previous study inferred that imatinib might be an effective drug for KD. However, further studies with actual clinical data are required for imatinib to be considered as a treatment option for KD [1,54].
Radiotherapy
Radiotherapy is considered an appropriate option for recurrent cases or poor surgical candidates. Hareyama et al. [55] reported that a 90% local control rate was achieved by using 26–30 Gy for irradiation and stated that the radiation field should be confined to the lesion and regional lymph nodes [55,56]. However, other reports showed that surgical excision combined with postoperative radiotherapy achieved a much lower local recurrence rate than surgery or radiotherapy alone [57]. Therefore, postoperative radiotherapy may be effective in controlling the residual lesion, reducing the recurrence rate without notable side effects.
Surgical excision
Surgery may allow the greatest diagnostic accuracy and has been the mainstay of treatment. However, as KD tends to be ill-defined on pathological examinations, it is difficult to achieve negative margins during surgical excision, which leads to a relatively high (25%) reported recurrence rate; furthermore, it was revealed that positive margins were a risk factor for disease recurrence [42,58,59]. Therefore, in clinical practice, surgical resection combined with low-dose postoperative radiotherapy has become the main treatment modality for KD.
Other interventions
In addition to conventional therapies, many other treatment options have been suggested. Suplatast tosilate, an anti-allergy drug, attenuated hyper-IgE and eosinophilia, suggesting that other mechanisms than the T cell-mediated immune response are related to the pathogenesis of KD. Cetirizine, which is another antiallergic agent, also showed beneficial effects in KD patients [60,61]. Boulanger et al. [62] reported that all-trans retinoic acids in combination with steroid therapy led to a rapid disappearance of clinical symptoms of KD. It was postulated that all-trans retinoic acids might exert immunomodulatory effects on Th2 cytokines and inhibit IL-4-mediated IgE production, causing the symptoms to disappear. A case report stated that sleep apnea due to KD improved with combination therapy of laser surgery, a steroid, and oral administration of pranlukast, which is a leukotriene receptor blocker [26]. Chewable nicotine also showed a treatment effect in skin disorders with eosinophilic infiltration [63]. A pediatric patient who had poor adherence to oral medications was treated with intravenous vincristine, resulting in remission of nephrotic syndrome complicated by KD and submandibular swelling [64]. However, these unconventional treatment options should be supported by large-scale clinical evidence, which requires further research.
PROGNOSIS
Even though a high overall recurrence rate (25%) has been reported with current treatment options–surgical excision (30.5%), medication (45%), and radiotherapy (60%)–KD has a favorable prognosis with no potential for malignancy. Sakamoto et al. [14] found that the eosinophil count was related to the therapeutic response in patients with KD and suggested that the eosinophil count might be used as a predictive marker for KD. According to other studies, patients who developed recurrence after surgical excision tended to have a maximal tumor diameter ≥3 cm, disease duration ≥5 years, peripheral eosinophil count ≥20%, or a serum IgE level ≥10,000 IU/mL, which required adjuvant therapy combined with surgical excision [2,57].
CONCLUSION
KD is a rare chronic inflammatory disorder with unknown etiology that frequently involves the subcutaneous tissue of the head and neck region. It is usually accompanied by regional lymphadenopathy and salivary gland involvement in Asian males. The distinct histologic features of KD include follicular hyperplasia with eosinophilic infiltration and preserved nodal architecture, making the diagnosis clear. Surgical resection combined with low-dose postoperative radiotherapy achieves the lowest local recurrence rate, and additional studies of immunosuppressive medications are required.