Epithelioid sarcoma associated with neurofibromatosis type I
Article information

Abstract
In general, patients with neurofibromatosis type I have a higher risk than those with other types of neurofibromatosis of developing soft-tissue sarcomas related to the nervous system. We here present a 42-year-old man with neurofibromatosis type I who developed a protruding mass over only 2 weeks. The histopathological diagnosis was epithelioid sarcoma. Epithelioid sarcomas are rare and, to the best of our knowledge, no epithelioid sarcomas have been reported in patients with neurofibromatosis type I. Radical excision of the primary lesion was performed and postoperative radiotherapy and chemotherapy administered, as is recommended for epithelioid sarcoma. Our case emphasizes that patients with neurofibromatosis type I may develop malignant tumors
INTRODUCTION
Neurofibromatosis type I (NF I) or von Recklinghausen disease, is an autosomal dominant disorder caused by inactivating mutations in the germline of the NF I gene. Characteristic features include pigmented skin lesions (freckling, cafe-au-lait spots), Lisch nodules, malignant or benign tumors, and organ or tissue damage (visceral, skeletal, and neural). Large complete gene deletions are associated with a high risk of development of malignant tumors of the nervous system, such as schwannomas, gliomas, and malignant peripheral nerve sheath tumors (MPNSTs). In particular, from 8% to 13% of patients with NF I develop MPNSTs [1-3]. However, NF I fibromatosis is generally benign and transformation into malignancy is rare. Also, malignant tumors unrelated to the nervous system rarely develop in patients with neurofibromatosis. However, few cases of NF I associated with sarcoma have been reported, including pleomorphic sarcoma [4], rhabdomyosarcoma [5], leiomyosarcoma [6], and Ewing sarcoma [7]. Here, we describe an unusual case of epithelioid sarcoma in a patient with NF I.
CASE REPORT
A 42-year-old man with a history of NF I presented with a growing mass on his posterior neck that he had first noted about 2 weeks earlier. Numerous hyperpigmented skin macules were observed on his back, abdomen and chest, and multiple cafe-au-lait spots on his shoulders and abdomen. Several neurofibromas were identified on his neck and abdomen (Fig. 1). He reported that his father and uncle had neurofibromas and cafe-au-lait spots on their skin.

Photographs showing cafe-au-lait spots on the patient’s right shoulder (A) and several neurofibromas on his neck (B).
The initial examination showed a 3.5 × 3.5 cm protruding mass with ulcerative bleeding on the posterior neck (Fig. 2). Malignancy was suspected because of the lesion’s rapid growth and surface ulceration. Because of the time required for biopsy, the patient refused to undergo biopsy first and instead requested immediate resection of the tumor. Accordingly, the mass was excised under general anesthesia and frozen sections examined. Excision was performed to the supramuscular layer with a 2 cm margin. Intraoperative examination of frozen sections resulted in a diagnosis of high-grade sarcoma with epithelioid features. After confirming that the resection margin was clear, the defect was covered by primary closure (Fig. 3). Pathological examination of the excised lesion confirmed the frozen section diagnosis of high-grade epithelioid sarcoma; the resection margin was clear. Immunohistochemistry studies showed that the tumor cells were strongly positive for cytokeratin (Fig. 4), but negative for CD34 and S100. They also showed weak nuclear expression of integrase interactor 1. Postoperatively, the patient underwent positron emission tomography-computed tomography (PET-CT) and enhanced neck CT; subcutaneous nodular lesions with enhancement were detected. Abnormal uptake of fluorodeoxyglucose was also detected in the lateral neck region bilaterally (Fig. 5). We therefore planned more extensive excision and sentinel lymph node biopsy with radical neck dissection and postoperative radiotherapy and chemotherapy. However, the patient consented only to excision of the suspicious lesions detected on radiologic evaluation. During the second operation on both lateral neck regions, abnormal tissue was found in the subdermal layer and excised with a clear resection margin. The excised lesions were diagnosed as epithelioid sarcomas. The patient agreed to postoperative therapy to minimize the risk of tumor metastasis. During a 6-month follow-up, he underwent radiotherapy with chemotherapy (doxorubicin and ifosfamide). No recurrence has been detected to date.

Photograph showing an approximately 3.5×3.5 cm protruding ulcerated mass with bleeding on the patient’s posterior neck.

Postoperative photograph.
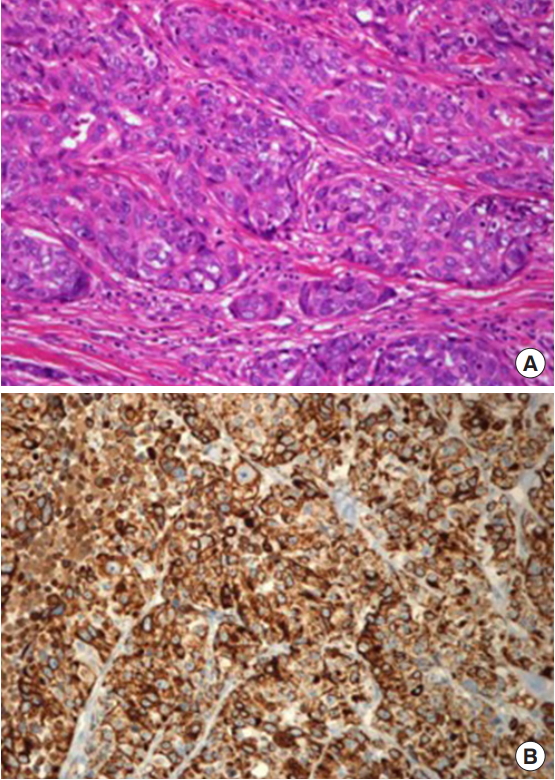
(A) Photomicrograph of the operative specimen showing nests of tumor cells with epithelioid features in the dermis (H&E, ×200). (B) Immunohistochemistry study showing that the tumor is strongly positive for cytokeratin (×200).
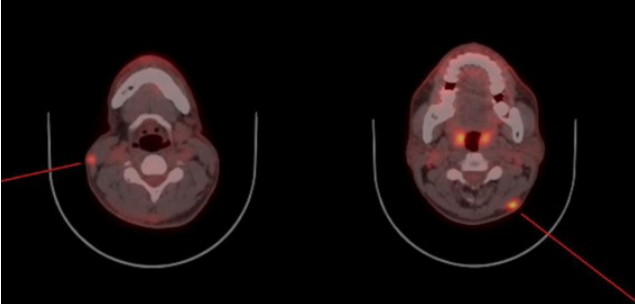
Positron emission tomography-computed tomography images showing fluorodeoxyglucose uptake in subcutaneous lesions.
DISCUSSION
NF I, an autosomal dominant disorder, is characterized by the development of multiple peripheral nerve sheath tumors called neurofibromas. There are seven diagnostic criteria for NF I according to the National Institutes of Health (Table 1) [8], two or more of which must be met to make a diagnosis of NF I. Neurofibromas and cafe-au-lait spots are benign and generally do not need to be treated. However, plexiform neurofibromas have the potential for malignant development. There is an 8% to 13% risk of developing malignant nerve sheath tumors. These malignant tumors are treated with extensive local resection; additionally, imatinib has been shown to reduce the size of plexiform neurofibromas [9]. Monitoring for neurologic changes is important because such changes may indicate tumor development. Although neurofibromas are benign, it has been well established that patients with NF I have a higher than general risk of developing soft-tissue sarcomas related to the nervous system [1,10-12]. Optic glioma is the most common central nervous system tumor associated with NF I [13], whereas malignant nerve sheath tumors are the most common peripheral nervous system tumor associated with NF I [14]. In some cases, NF I malignant transformation is associated with heterologous differentiation of components [4], commonly including angiosarcoma, chondrosarcoma, rhabdomyosarcoma, and osteosarcoma.

Diagnostic criteria for neurofibromatosis type I
Epithelioid sarcoma, first described by Laskowski in 1961 [15], is a rare soft-tissue tumor with a propensity for local recurrence, regional lymph node involvement, and distant metastasis. It commonly presents as a slow-growing, firm-to-hard palpable mass with surface ulceration and invasive borders and disseminates via subdermal lymphatic vessels [16]. After the establishment of histopathological diagnosis, epithelioid sarcomas are treated by radical excision. Sentinel lymph node biopsy with therapeutic lymph node dissection is indicated in patients suspected of having regional lymph node or systemic metastases. Radiation can also be considered and adjuvant chemotherapy is indicated when metastatic disease is suspected or confirmed [17-20].
There is currently no known direct association between neurofibromatosis and epithelioid sarcomas. To the best of our knowledge, this is the first report of an epithelioid sarcoma developing in a patient with NF I. We did not establish a direct association between the above two diseases in our patient. However, considering that patients with neurofibromatosis are at high risk of developing multiple malignancies, it would be of interest to compare our patient’s disease course with that of patients with NF I and the more commonly associated malignant tumors. The present case highlights that physicians should be aware of the possibility of malignant tumors in patients with NF I, as well as their histological characteristics and prognoses.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Ethical approval
The study was performed in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained.
Patient consent
The patient provided written informed consent for the publication and the use of his images.