Current concepts of neurofibromatosis type 1: pathophysiology and treatment
Article information

Abstract
Neurofibromatosis type 1 is the most common tumor predisposition syndrome inherited in an autosomal dominant (100% penetrance) fashion with a wide variety of expressivity. From the perspective of plastic surgery, the most significant clinical symptoms, including disfiguration, peripheral neurologic symptoms, and skeletal abnormalities, are caused by various tumors originating from the affected nerves. Surgical removal is the standard of care for these tumors. However, the outcome is frequently unsatisfactory, facilitating the search for additional therapeutic adjuvants. Current trials of molecularly targeted therapies are promising.
INTRODUCTION
Neurofibromatosis (previously known as von Recklinghausen disease) are distinct clinical entities that represent numerous neurofibromas on the skin. Neurofibromatosis type 1 (NF1; OMIM:162200) is characterized by multiple café-au-lait macules (CALMs) and cutaneous neurofibromas (CNs). It frequently involves the head and neck region and exhibits a variety of symptoms, ranging from simple skin neurofibromas to devastating plexiform neurofibromas (PNs) that cause melting-skin disfiguration, blindness, nerve compression, and airway obstruction (Fig. 1) [1].

Optic nerve glioma arises in the optic nerve. Blindness may occur in approximately 5% of the patients.
This article reviews the fundamental features of NF1 and current knowledge and individual strategies for disease management, particularly in the head and neck region.
HISTORY
NF1 was first described as a systemic disorder by Friedrich von Recklinghausen in 1882 [2]. Early studies focused on its clinical presentation and hereditary nature. This led to the development of a diagnostic criteria consensus in 1987 by the National Institutes of Health (NIH) [3]. Specialized clinical investigations have helped to recognize various neurodevelopmental symptoms other than the tumorigenic presentation of the disease [4].
Recently, the NF Clinical Trials Consortium was established to aid biologic experimental studies and the Response Evaluation in Neurofibromatosis and Schwannomatosis to collect clinical outcomes more uniformly [5-7].
There is an expansion in the knowledge of clinical disorders caused by germline mutations in genes encoding products of the Ras/mitogen-activated protein kinase (MAPK) pathway. These are considered RASopathies, and now, NF1 is regarded as the first one identified [8].
EPIDEMIOLOGY
NF1 is the most common form of neurofibromatosis, with an estimated incidence of 1 in 2,700 and a prevalence of 1 in 4,500. NF1 represents monogenic disorders presenting a pattern of autosomal dominant inheritance, with a strikingly high rate of de novo mutations that were found to be 42%. These de novo mutations are believed to be responsible for the sporadic appearance of NF1 [9]. Penetrance is complete or, at least, nearly so before the age of 5 years, but expressivity differs greatly between affected individuals, even within the same consanguinity. There is no predilection for sex, race, or ethnicity [10,11].
The lifetime risk of malignancy in individuals with NF1 is estimated to be 59.6% [12]. This justifies the lifelong surveillance of patients with NF1 [13].
GENETICS, MOLECULAR PATHOPHYSIOLOGY, AND GENOTYPE-PHENOTYPE CORRELATIONS
NF1 occurs as a result of a heterozygous germline mutation in the NF1 tumor suppressor gene (HGNC:7765). NF1 is located on the long arm of chromosome 17 (17q11.2) and carries a genetic code for the production of neurofibromin [14,15]. Neurofibromin, a 220-kDa cytoplasmic protein, is an important negative regulator of Ras, a proto-oncogene that plays a key role as a signaling molecule in cell growth and differentiation. Ras mutation is associated with a wide variety of cancer types and has been identified in a series of clinical entities called RASopathies [16].
Neurofibromin is a ubiquitous protein produced by most cells, particularly at high levels in cells of the nervous system (neurons, oligodendrocytes, and non-myelinating Schwann cells). Astrocytes and myelinating Schwann cells do not produce neurofibromin. This finding may explain some of the clinical manifestations, such as nerve tumors and cognitive disabilities [17].
Every affected individual carries a defective NF1 allele (one of paired arms). The other copy of NF1 on the other allele is considered responsible for producing functional neurofibromin. Additional loss-of-function mutations in the functioning NF1 copy may result in a wide variety of symptomatic presentations. These somatic mutations occur during embryonic development, resulting in mosaicism of the NF1 copies. Mutations in the early stages cause more generalized symptoms resembling non-mosaic NF1 due to germline mutations. Later in development, mutations in more differentiated cells manifest as more localized symptoms, often described as segmental NF1 [18]. This “2-hit” theory was suggested to explain the development of several NF1-related tumors, including malignant peripheral nerve sheath tumors (MPNSTs). A phenotype is a result of the complex interplay between multiple factors. Links between genetic impairments and clinical presentation are still under investigation. More than 3,000 different pathogenic sequence variants have been identified; however, only four genotype-phenotype correlations have been clinically identified [7]. An in-frame deletion of codon 992 (p.Met992del) and a missense mutation in codon 1809 (p.Arg1809Cys) are associated with the absence of CNs or PNs [19-21]. Other missense mutations affecting one of codons 844–848 and some submicroscopic chromosomal deletions, so-called microdeletions, are associated with a more severe phenotype [22,23]. Current knowledge for genotype-phenotype correlations is limited because of the genetic heterogeneity and absence of mutational “hot spots” [7].
At present, NF1 is diagnosed using established diagnostic criteria. Genetic testing is reserved for tricky clinical presentations and reproductive decision-making.
DIAGNOSIS
The US NIH Consensus Development Conference proposed clinical diagnostic criteria for NF1. Seven criteria were suggested, of which at least two criteria are required to confirm the diagnosis (Table 1) [3].

Conventional diagnostic criteria of NF1
However, some clinical manifestations of NF1 are age-dependent (i.e., neurofibromas take time to become evident). Therefore, diagnosis is not easy in early childhood particularly for sporadic NF1 cases (because they have no parents with NF1). Approximately 46% of patients fail to meet the diagnostic criteria by the age of 1 year. Close persistent monitoring can reveal the hallmark symptoms of NF1, which are included in diagnostic criteria. In 97% of children suspected of having NF1, the criteria were met by the age of 8 years.
Virtually all suspected cases are diagnosed by the age of 20 years. The order of appearance of the skin manifestations was CALMs, axillary freckling, Lisch nodules, and CNs or subcutaneous neurofibromas or PNs. Thus, CALM is often the first symptom that suggests the penetrance of NF1 [24]. CALMs tend to increase in size and number as the patient grows. By adulthood, approximately 95% of patients with NF1 will have light-brown skin hyperpigmentation lesions [25]. Genetic testing is not routinely recommended [7].
Recently, an international consensus group has suggested revised diagnostic criteria. Major changes included new separate diagnostic criteria for mosaic NF1 and new criteria for genetic diagnosis. The group recommended performing genetic analyses for patients with segmental symptoms, families with two or more affected siblings and unaffected parents, and children presenting only pigmentary symptoms. Sphenoid wing deformation with an anatomically relevant PN is considered insufficient for sphenoid wing dysplasia [26].
CLINICAL FEATURES AND MANAGEMENT
Cutaneous neurofibromas
CNs are the most common type of skin tumors in NF1. CNs present as well-defined cutaneous lesions that are localized but not encapsulated (Fig. 2) [27]. The prevalence of CN in patients with NF1 reached up to 99% [2]. They are benign tumors composed of atypical Schwann cells but also contain fibroblasts, mast cells, pericytes, and perineural cells [28-33]. CNs arise within the epidermal/dermal layer and increase in size and number after an appearance in late childhood [34]. CNs tend to grow more rapidly during puberty and pregnancy, but discrete evidence from prospective observations is lacking [35,36].
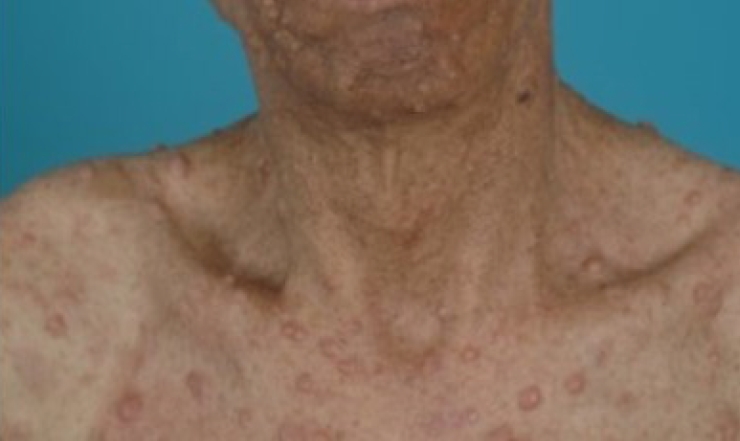
Cutaneous neurofibromas are presented as multiple, well-defined cutaneous lesions.
The shape of a CN is classified into the following five categories: nascent/latent, flat, sessile, globular, or pedunculated [37]. Approximately 20% of patients experience pruritus related to CNs [38]. The pathophysiology of pruritus in NF1 is still not clearly elucidated, but some studies suggest that mast cells may play a significant role [39-41].
Diffusely infiltrating PNs should be considered in the differential diagnosis since these PNs arise from deeper tissues and mimic CNs by dermal invasion [42]. Any atypia seen on a smear of a PN specimen has the possibility of early malignant transformation, whereas CN specimens often appear to show atypical cells reflecting reactive or degenerative changes [43]. Although CNs are benign and carry no risk of malignant transformation, they can still be troublesome because hundreds to thousands of CNs can cause significant disfigurement, particularly when they involve the head and neck region, causing emotional and physical discomfort [44].
The primary treatment for CNs is surgery. Excision is effective enough to remove one tumor; however, given that CNs are frequently numerous in number and size, excision is not feasible for the management of entire skin lesions [45,46].
Plexiform neurofibromas
PNs are a distinct feature of NF1 that causes severe clinical discomfort. They are histologically benign but can be devastating clinically and aesthetically. The prevalence of PNs among patients with NF1 is up to 50% [47,48].
PNs differ from CNs in that they can involve multiple fascicles and nerve branches [49]. They can cause significant discomfort to patients, such as pain, disfiguration, mass effect, and functional impairments of neurovascular structures and even the airway [50]. PNs are also believed to cause increased mortality owing to their risk of malignant transformation to MPNSTs throughout their lifespan [51,52]. The risk of malignant transformation of PNs to MPNSTs is increased 20-fold [53], reaching a 10%–15% lifetime risk.
PNs may be congenital, and their growth has been described as unpredictable [49]. Recent studies have found that the growth of PNs bursts out rapidly during childhood and adolescence, and then the rate diminishes over time to converge to zero growth in young adulthood [54]. Thus, unusual growth of PNs or any accompanying neurologic symptoms due to PNs in adulthood may be a warning sign of existing malignancies; therefore, active evaluation and careful monitoring are required. Magnetic resonance imaging (MRI) is the gold standard for the diagnosis and monitoring of PNs [47,54]. However, treatment decisions are based on clinical presentation. Thus, whether, when, and where to image targeted PNs are done as per the practitioners’ judgments [55].
Diffuse PNs appear to infiltrate tissues adjacent to the involved peripheral nerves; therefore, they are not well demarcated (Fig. 3). In contrast, nodular PNs show well-demarcated margins (Fig. 4) [56].
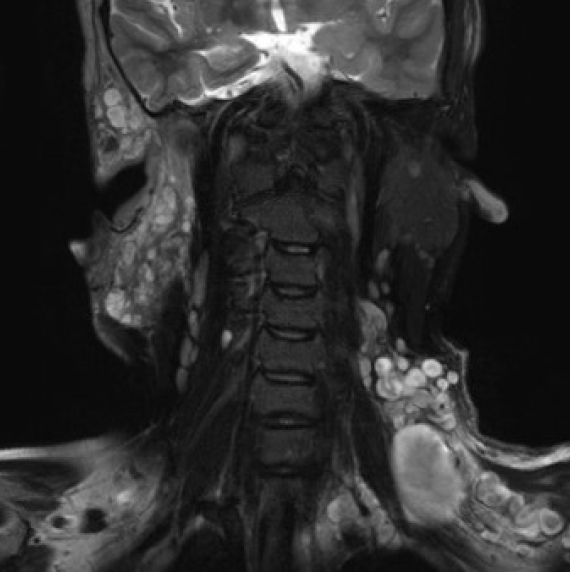
Diffuse plexiform neurofibromas infiltrate adjacent tissues and involve peripheral cervical nerve branches. They are not well demarcated.
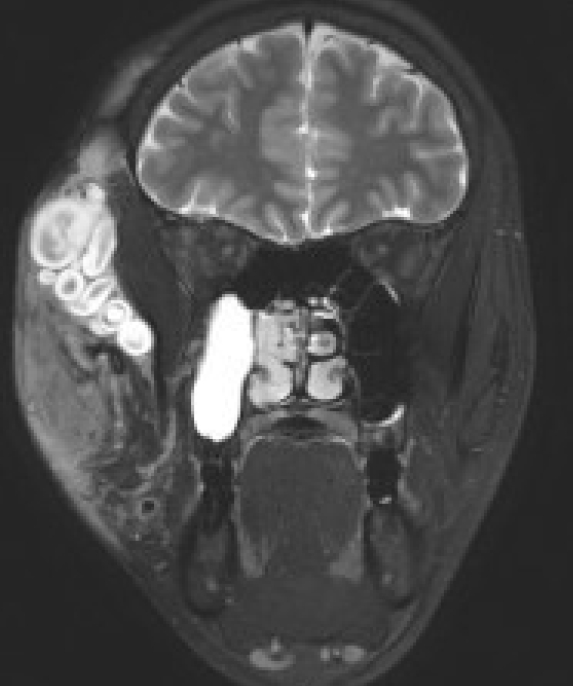
Nodular plexiform neurofibromas, originating from the trigeminal nerve (zygomaticotemporal nerve of V2), exhibit well-demarcated margins.
The only standard treatment for PNs is complete surgical removal, and indications for surgical removal are primarily symptomatic, including pain, severe disfiguration, and loss of neurologic and aesthetic function [13]. However, it is difficult to achieve an ideal result because of the large size, location, infiltration to adjacent tissue, and eventual recurrence of PNs. Furthermore, PNs, large enough to be symptomatic, are frequently accompanied by the laxity of the covering skin, risk of severe bleeding from impaired blood vessels, and poor separation from the nerve fibers [31,57]. The skin area involved with underlying PNs loses the elasticity, which results in the absence of normal viscoelastic properties of the skin, such as mechanical creep and stress relaxation [58].
In surgical cases, an intraoperative nerve stimulator and/or an operative microscope will help clearly distinguish the lesion from the surrounding tissue (Fig. 5). Preoperative angiography and/or percutaneous vascular embolization may be performed to prevent intraoperative bleeding from the surrounding tissue as in the treatment of vascular malformations [59]. However, the tumor microenvironment (TME) frequently contains weak blood vessels with dysplastic walls, which are prone to cause bleeding and difficult hemostasis. Since ectatic and dysplastic veins and capillaries are the main foci of bleeding, arterial embolization frequently has a limited effect.
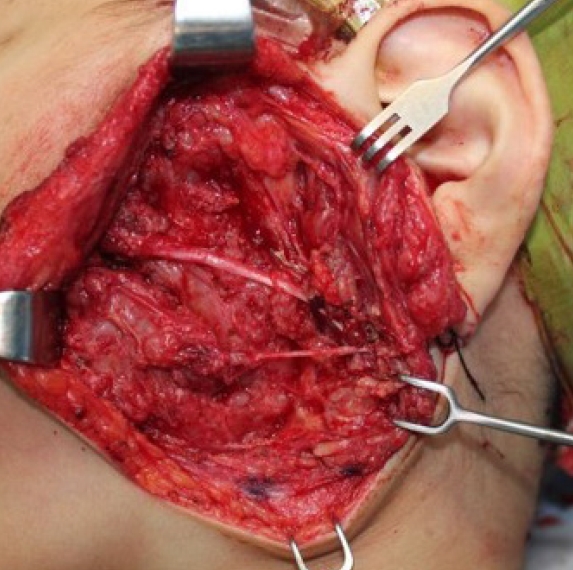
Dissection and preservation of facial nerve branches using an intraoperative nerve stimulator while resecting a plexiform neurofibroma of the cheek.
In the head and neck, rich vascular collateralization and shallow positioning of major vessels make hemostasis even more difficult. The vessels in NF1 seem to lack functioning tunica media in the wall, so bleeding from the stretched vessels tends to be low-pressure bleeding, and electrocautery, in this case, does not work well as there is weak or no contraction of the muscle layer. Therefore, physical ligation of the vessels using sutures or external compression is required to achieve appropriate hemostasis. Surgeons should be aware of the risk of intractable bleeding as the resection plane becomes deeper and closer to the neck. Surgical removal of PNs is often performed as palliative rather than curative care, with the goal of symptomatic relief rather than margin-free extirpation. In this circumstance, active surveillance is needed because PN tends to regrow within a few years.
PNs resistant to surgical management are candidates for modern targeted therapies, which will be reviewed in the following section.
Malignant peripheral nerve sheath tumors
MPNST is a subtype of malignant soft-tissue tumors (i.e., sarcomas). According to the World Health Organization classification system, it is presumed to be originated from Schwann cells. The trunk and extremities are most frequently involved, whereas 15% of the cases involve the head and neck region [60]. MPNSTs occur in 8%–16% of patients with NF1 and exhibit poorer outcomes than in those without NF1 [12,61]. They are the most common cause of mortality in patients with NF1, showing 15%–50% of a 5-year survival rate [61-64]. These high-grade tumors are associated with a high frequency of distant metastasis and regional recurrence [65].
Several risk factors for developing MPNST include young age [53], the whole-body burden of internal PNs [56,66], presence of pain [67], presence of nodular or atypical neurofibromas [43,68], prior radiation therapy [61], family history of MPNST [69], and microdeletion of the NF1 gene [70].
MPNSTs can occur sporadically but often develop within preexisting PNs [43,71]. MRI can help determine the location and extent of the tumor, which is seen as peripheral enhancement, peripheral edema, intra-tumoral cyst formation, necrosis, and heterogeneity (Fig. 6) [72]. 18F-fluoro-deoxy-glucose (FDG)-positron emission tomography (PET) has been shown to have better sensitivity for distinguishing malignancies from benign lesions, given that an MPNST will uptake more of FDG than a basal level (Fig. 7) [63,64]. Functional MRI and FDG-PET, together, showed a high sensitivity for detecting malignant transformation. Thus, they can help direct surgical intervention [73,74].
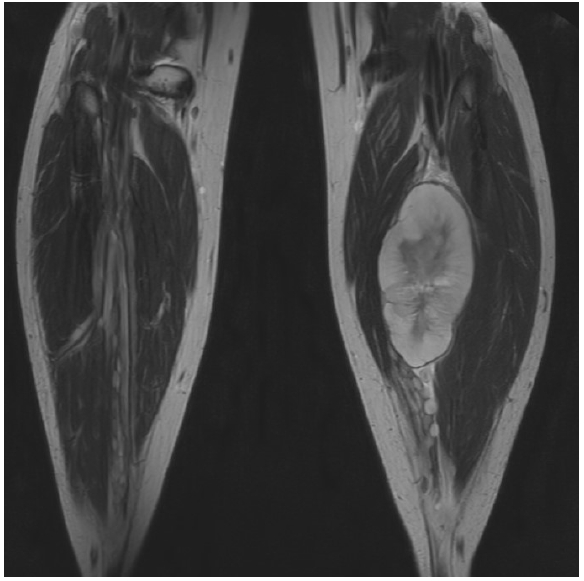
A malignant peripheral nerve sheath tumor, occurring within the tibial nerve, shows peripheral edema, intra-tumoral cyst formation, necrosis, and heterogeneity.

A positron emission tomography scan shows a large malignant peripheral nerve sheath tumor in the left cheek of the patient who has neurofibromatosis type 1 involving the entire face.
Surgery is the only curative treatment option. This is correct for localized disease if the surgery is performed in the early stages because the tumor is resected completely without neurologic damage and a safe margin is achieved. However, this is often limited because MPNSTs frequently invade adjacent structures and involve vital neurovascular networks [75].
Nonetheless, complete resection and tumor-free margins are important indicators for better prognosis [76], whereas old age, metastatic lesions at the time of diagnosis, and only biopsy without resection were significant and independent predictors of poor outcomes [77].
Radiation therapy is generally avoided in patients with NF1 owing to their predisposition to cancer development; however, it can still be used for limited purposes, including local control, especially in cases of incomplete tumor resection. Radiation appears to delay recurrence without affecting mortality [63]. However, further investigation is needed since the association between radiation therapy and mortality, especially after oncologic resection and reconstruction for scalp lesions, has been reported [78].
The efficacy of chemotherapy remains controversial [79,80]; however, in advanced stages, a single-agent anthracycline or doxorubicin-ifosfamide dual regimen can be used for palliative purposes [81,82]. Recently targeted therapies are reviewed in the following section.
Morphologic changes of facial bones
The most distinctive bony abnormality of NF1 in the field of plastic surgery is sphenoid wing dysplasia, with a prevalence of 11.3% in the probands (Fig. 8) [83]. The sphenoid wing dysplasia typically occurs unilaterally and involves the affected-side orbital walls causing orbital fossa widening, expansion of middle cranial fossa, herniated temporal lobe, and pulsating exophthalmos. Consequently, the malar and zygomatic arches are displaced downward [84].
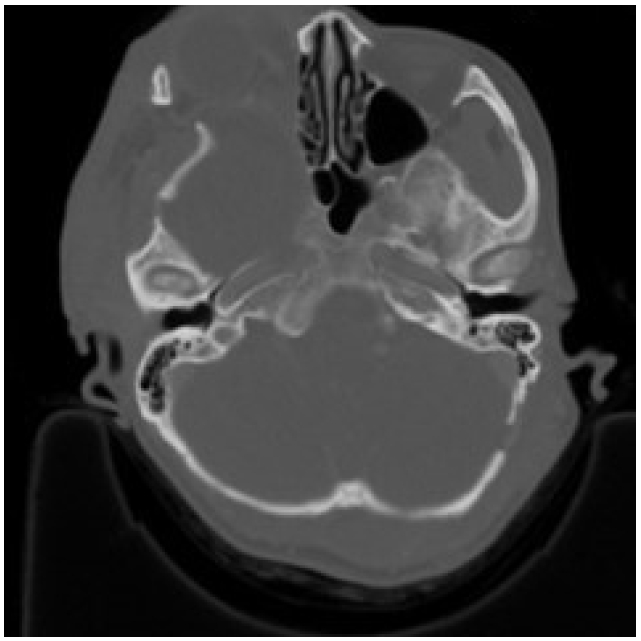
A computed tomography scan shows sphenoid wing dysplasia and orbital wall defects.
Sphenoid wing dysplasia is associated with an anatomically relevant space-occupying lesion, which is mostly a PN that causes local compression. Most patients do not require bone correction [85]. However, if there are symptomatic presentations, removal of the tumor is required. Flaps can be utilized to fill the resultant dead space, which is further accentuated by the dysplastic sphenoid bone [86].
Mandibular dysplasia can also be seen in patients with NF1 and is characterized by a pathognomonic deep sigmoid notch (Fig. 9) as well as a wide inferior alveolar canal, enlarged mandibular foramen, and notching of the inferior border (antegonial notch) [87]. The clinical significance and management strategies for mandibular dysplasia are not clear in the literature.
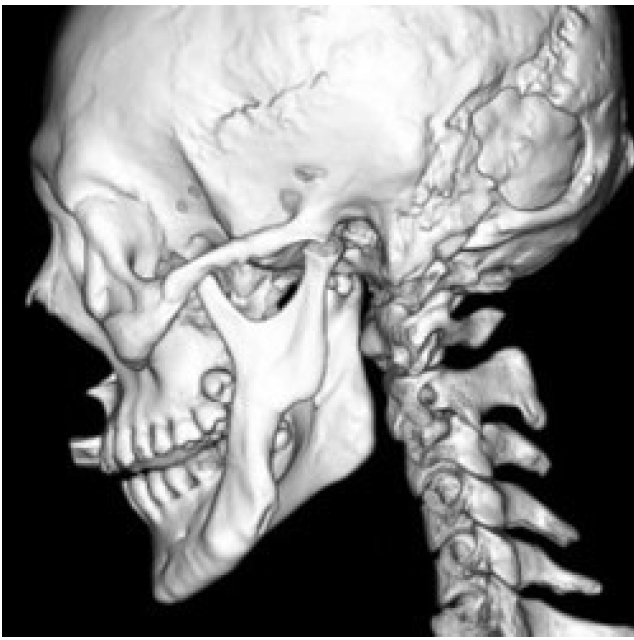
A 3-dimensional computed tomography scan shows a deep sigmoid notch, which is pathognomonic, as well as an increased antegonial notch size.
MOLECULAR TARGETED THERAPIES
Surgery is the current standard of care for both benign and malignant tumors in patients with NF1. However, because tumor recurrence after surgery is not rare, the need for adjuvant therapy has been proposed [88]. Early trials of general agents, including anti-inflammatory, antifibrotic, and antiangiogenic agents, failed to achieve therapeutic significance in the management of PNs [89-91]. A more recent trial with pegylated interferon showed that it elongates the time to progression. However, the application of interferon therapy is limited owing to side effects [92].
The cumulative knowledge of the molecular pathophysiology of PNs has enabled the development of molecular targeted therapies. The protein product of the NF1 gene, neurofibromin, is a negative regulator of Ras protein, and defective NF1 produces dysfunctional neurofibromins, resulting in constitutive activation of the Ras pathway and, thus, tumorigenesis [93]. Ras activates RAF kinase, MEK, and MAPK (ERK). MAPK regulates the transcription factors involved in the cell cycle [16].
Selumetinib, a selective MEK inhibitor, was the first medication approved by the US Food and Drug Administration for the treatment of intractable, symptomatic PNs in children aged > 2 years. It has been proven to decrease the volume of PNs for more than 1 year even with symptomatic relief [94,95]. Owing to the success of selumetinib, other MEK inhibitors, including trametinib, binimetinib, and mirdametinib, have been investigated, and, so far, the trials have shown remarkable success [96].
Other advancements in molecular targeted therapy of PNs were achieved by studies of cabozantinib, a tyrosine kinase inhibitor that plays a critical role in the regulation of the TME. In a phase II study, 42% of patients achieved partial response, which indicates a > 20% reduction in tumor volume [97].
Most clinical trials investigating targeted therapy regimens for MPNST are still ongoing.
CONCLUSION
NF1 is the most common tumor predisposition syndrome inherited in an autosomal dominant (100% penetrance) fashion with a wide variety of expressivity. From the perspective of plastic surgery, the most significant clinical symptoms, including disfiguration, peripheral neurologic symptoms, and skeletal abnormalities, are caused by various tumors originating from the affected nerves. Surgical removal is the standard of care for these tumors. However, the outcome is frequently unsatisfactory, facilitating the search for additional therapeutic adjuvants. Current trials of molecularly targeted therapies are promising.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Ethical approval
The patients provided written informed consent for the publication and the use of their images.
Author contribution
Conceptualization: So Young Lim. Data curation: Jaemin Choi, Sungbin An. Writing - original draft: Jaemin Choi, Sungbin An, So Young Lim. Writing - review & editing: Jaemin Choi, Sung-bin An, So Young Lim. Supervision: So Young Lim.
Abbreviations
CALMs
café-au-lait macules
CNs
cutaneous neurofibromas
FDG
18F-fluoro-deoxy-glucose
MAPK
mitogen-activated protein kinase
MPNSTs
malignant peripheral nerve sheath tumors
MRI
magnetic resonance imaging
NF1
neurofibromatosis type 1
NIH
National Institutes of Health
PET
positron emission tomography
PN
plexiform neurofibromas
TME
tumor microenvironment