Abbreviations:
AVM,
arteriovenous malformation
;
BRBNs,
blue rubber bleb nevus syndrome
;
CM,
capillary malformation
;
GLUT1,
glucose transporter isoform 1
;
ISSVA,
International Society for the Study of Vascular Anomalies
;
KH,
kaposiform hemangioendothelioma
;
KTS,
Klippel-Trenaunay syndrome
;
MRA,
magnetic resonance angiography
;
MRI,
magnetic resonance imaging
;
SWS,
Sturge-Weber syndrome
;
TA,
tufted angioma
;
VEGF,
vascular endothelial growth factor
.
INTRODUCTION
Vascular anomalies encompass a variety of malformations and tumors that can result in severe morbidity and mortality in both adults and children. Progress has been made in the classification and diagnosis of these anomalies [1]. They are divided into two categories: vascular tumors and vascular malformations. Vascular tumors result from the abnormal growth of blood vessels, whereas vascular malformations are structural abnormalities that arise during embryonic development, impacting either blood vessels or lymphatic vessels. Examples of vascular tumors include infantile hemangioma, kaposiform hemangioendothelioma (KH), and tufted angioma (TA). In contrast, vascular malformations consist of capillary, venous, lymphatic, and arteriovenous malformations (AVMs). The diagnosis and management of vascular anomalies can be challenging due to their variable presentations and complex underlying mechanisms [2,3]. However, advances in diagnostic techniques and treatment options have led to improved outcomes for patients with these conditions [4].
CLASSIFICATION
The International Society for the Study of Vascular Anomalies (ISSVA) classification system is a widely accepted approach employed by healthcare professionals who specialize in treating patients with vascular anomalies. This system involves the use of histopathology, clinical presentation, and treatment options to categorize these malformations [5]. In 1982, Mulliken and Glowacki [6] introduced an organizational system for vascular anomalies based on their pathological characteristics, dividing them into binary groups. Similarly, the ISSVA classification system catalogs vascular anomalies into two primary biological categories: vasoproliferative neoplasms (or vascular neoplasms) and vascular malformations. The primary difference between these two groups is the presence of increased endothelial cell turnover, which is identified by observing mitoses on histopathology [7]. Vasoproliferative neoplasms undergo mitosis and exhibit enhanced endothelial cell turnover. In contrast, vascular malformations do not exhibit augmented turnover. These structural abnormalities of the capillary, venous, lymphatic, and arterial systems grow in proportion to the child [7,8]. The use of the suffix “-oma” to describe both neoplasms and malformations is being abandoned. The term “lymphangioma” is considered a misnomer, while “hemangioma” requires a qualifier such as “infantile” or “congenital” to meaningfully describe distinct pathological lesions [9]. The ISSVA categorization system helps avoid confusion in the diagnosis and treatment of vascular malformations [10]. A study of children at a vascular malformation clinic indicated that the use of the ISSVA system reduced misdiagnoses from an initial rate of 69%; additionally, it decreased the proportion of parents receiving incorrect information about lesion progression and treatment from 53% [11].
DIAGNOSIS: IMAGING STRATEGIES FOR VASCULAR ANOMALIES
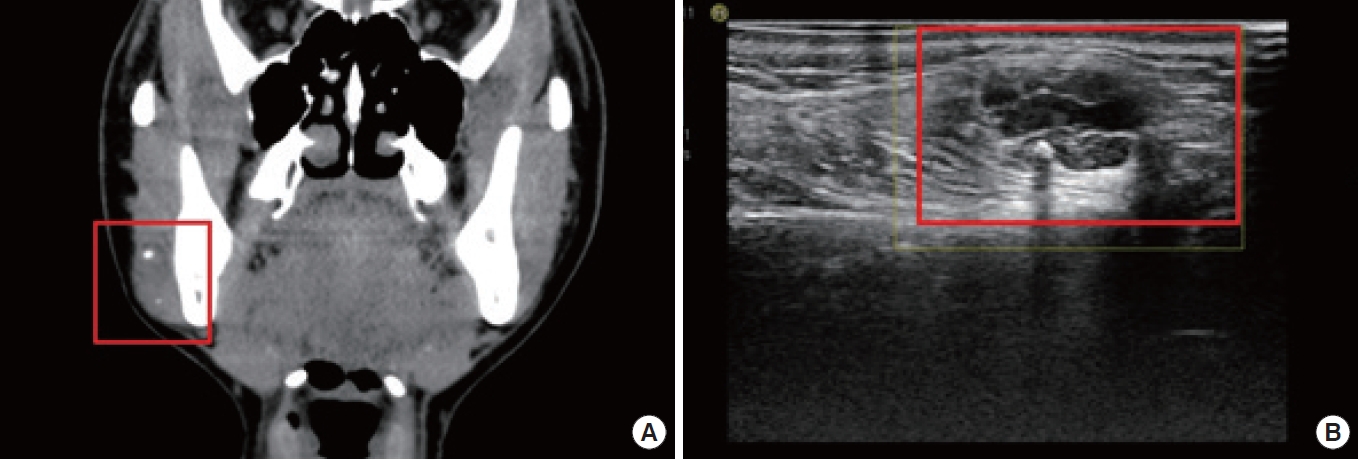
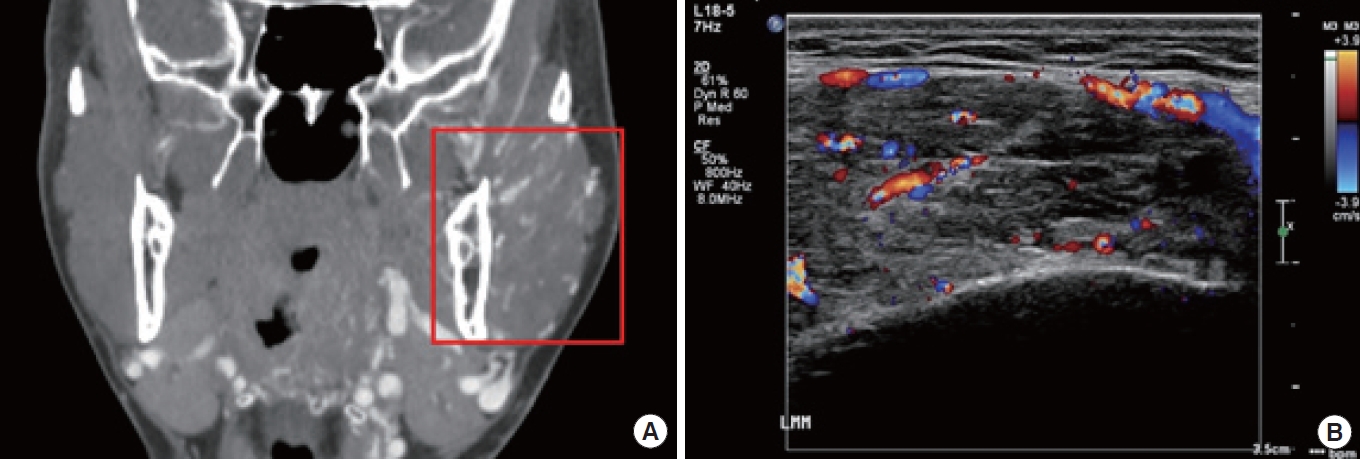
Vascular anomalies are often diagnosed through a patient history and physical examination, but imaging can also provide valuable insights [4]. The choice of imaging modality should depend on the specific lesion and clinical situation. Ultrasonography and magnetic resonance imaging (MRI) are the primary techniques used to identify and characterize vascular malformations and tumors. Contrast-enhanced ultrasound can be particularly helpful in visualizing lesions that are poorly defined (Fig. 1) [12]. High-frequency linear transducers are recommended for imaging skin lesions, whereas lower-frequency transducers may be necessary for deeper lesions. Lymphatic malformations can often be identified on ultrasound by the absence of blood flow within the lesion. The fluid within the cystic structures typically appears either echo-free or mildly echogenic. In the case of bleeding into the malformation or bacterial infection, the appearance of the lymphatic fluid on ultrasound may shift from hypoechogenic to hyperechogenic [13]. Color Doppler ultrasound provides a comprehensive assessment of fast-flow arteriovenous shunts, which are commonly accompanied by flow-related arterial, nidal, and/or venous aneurysms resulting from the deterioration of the dysplastic vessel wall. This non-invasive imaging technique provides valuable information for the diagnosis and management of arteriovenous anomalies (Fig. 2,3) [13]. On T1-weighted MRI sequences, non-thrombosed hemangiomas exhibit hypointensity relative to muscle, but they demonstrate marked hyperintensity with flow voids upon administration of contrast agent due to their pronounced vascular perfusion. This results in a characteristic appearance often likened to a “bag of worms” [14]. On T2-weighted images, venous malformations exhibit distinct and strong hyperintense signals. On pre-contrast T1-weighted sequences, venous malformations appear similar in intensity to muscular tissue. However, on post-contrast T1-weighted sequences, they display hyperintensity due to the accumulation of contrast agent within the lesion [13]. A typical finding on the MRI T2-weighted imaging of a macrocystic lymphatic malformation is a strong hyperintense signal within the lesion, while the cyst walls or septae show contrast enhancement on T1-weighted sequences (Fig. 4) [13]. Dynamic contrast-enhanced magnetic resonance angiography (MRA) and post-contrast T1-weighted isovolumetric gradient echo sequences are the preferred imaging techniques for demonstrating flow dynamics, inflow arteries, outflow veins, and the nidus location in AVMs [15]. To identify bone involvement in AVMs, post-contrast T1-weighted images are the most effective, as they illustrate the intensive contrast uptake of the intraosseous vessels. When evaluating fast-flow vascular anomalies, MRA is useful; this includes non-contrast imaging angiography using two-dimensional time-of-flight or phase-contrast imaging. Another technique is dynamic imaging with gadolinium-enhanced time-resolved MRA, which can separate the arterial and venous phases for imaging and help visualize feeding arteries, draining veins, and shunt locations [16]. These techniques are valuable in the assessment of vascular anomalies.
Computed tomography (CT) is employed when MRI is contraindicated, while other imaging modalities such as catheter angiography and direct percutaneous phlebography are used in peri-interventional circumstances. Molecular imaging using positron emission tomography/CT can be used to diagnose and differentiate certain vascular tumors [17]. Differentiating between high-flow and low-flow vascular anomalies is crucial in determining the appropriate management approach. Although plain radiographs are not typically used in diagnosis, they may offer insights in cases with nonspecific symptoms. X-rays can be utilized to evaluate any osseous alterations that may be associated with vascular anomalies. These changes typically arise from malformations rather than hemangiomas and include periosteal response, distinct radiolucent bone lesions, asymmetrical leg length, and overgrowth on the affected side [18]. Phleboliths that have calcified around a venous malformation may be identified, suggesting previous episodes of thrombophlebitis that have gradually calcified over time. This finding can be visualized on X-rays. Furthermore, accurate automatic segmentation of vascular tumors or malformations, including those in the head and neck region, may be a viable option to determine their extent and volume [19].
VASCULAR TUMORS
To avoid potential confusion, it is important to understand the nomenclature of vascular tumors, which often have similar terminology. The ISSVA classification system encompasses a variety of vascular tumors. These include infantile hemangioma, congenital hemangioma (such as rapidly involuting and noninvoluting congenital hemangiomas), TA, KH, spindle cell and epithelioid hemangioendotheliomas, and angiosarcoma.
Based on the available evidence, vasoproliferative tumors appear to originate from vasculogenesis-the formation of new blood vessels from angioblasts-and not angiogenesis, which is the growth and branching of existing blood vessels. This represents a departure from the earlier belief that these tumors arose primarily from angiogenesis [20].
Markers of cellular proliferation, including vascular endothelial growth factor (VEGF), are elevated in actively growing hemangiomas, but not in those that are involuting [21]. Moreover, the expression of glucose transporter isoform 1 (GLUT1) is a unique marker for placental endothelial cells and infantile hemangiomas, absent in all other normal tissues or vascular tumors [22]. Research has suggested that the life cycles of placental cells and infantile hemangiomas are similar, leading to speculation that hemangiomas may be the result of embolized placental cells [23].
Certain hemangiomas appear to exhibit a non-random, regional distribution that may be linked to embryological prominences, suggesting a potential neuroectodermal abnormality [24]. For instance, facial hemangiomas are frequently observed around embryologic fusion lines. This can be attributed to the fact that the mesenchyme in the fetal head and neck primarily consists of neuromesenchyme. This composition facilitates hemangioblast migration, which may clarify why hemangiomas frequently develop in the head and neck region [23]. Facial embryologic fusion lines are areas where embryonic tissue from two different facial regions fuses during fetal development. These lines may appear as faint ridges on the surface of the skin. The exact location of these fusion lines varies among individuals, but they typically occur around the eyes, nose, and mouth. Examples of facial embryologic fusion lines include the nasolacrimal groove (running from the inner corner of the eye to the nostril), the philtrum (the vertical groove between the nose and the upper lip), and the midline of the chin. Hemangiomas often occur in areas surrounding these fusion lines [25].
Infantile hemangioma
Infantile hemangioma, a benign tumor, is the most common tumor found in infancy. It is estimated to affect approximately 4% to 10% of infants, with higher incidence rates observed in female and premature infants [26]. Certain cellular proliferative markers, such as VEGF, are elevated in proliferating hemangiomas but not in involuting hemangiomas [21]. This suggests that VEGF may play a role in the growth and development of infantile hemangiomas. Another unique marker, the GLUT1 protein, is expressed in both placental endothelial cells and infantile hemangiomas. GLUT1 is not expressed by any other normal tissue or vascular tumor, making it a useful diagnostic marker for infantile hemangioma. The expression of GLUT1 in these tumors likely relates to the increased glucose uptake necessary for the rapid cellular proliferation that occurs during the tumor growth phase [22].
Infantile hemangiomas can appear anywhere on the body, but they are most commonly found on the skin. The head and neck are the most frequent locations, representing approximately 60% of cases, while the trunk and extremities account for 25% and 15% of cases, respectively [23]. Hemangiomas on the face, scalp, and neck are particularly concerning due to the risk of functional impairment or disfigurement. These cases may necessitate close monitoring and early intervention to prevent complications. In rare cases, hemangiomas can also occur in internal organs such as the liver, lungs, or brain, which may require specialized treatment.
Liver hemangiomas can be classified as focal, multiple, or diffuse, based on their size and distribution within the liver. The term “hemangiomatosis” has been used by some authors to describe the presence of multiple infantile hemangiomas, while “disseminated hemangiomas” may be used to refer to hemangiomas found in three or more organs [27]. However, for simplicity, the term “infantile hemangioma(s)” is generally used to describe any type of hemangioma in infants. While most infantile hemangiomas are benign and do not require treatment, those located in certain areas or exhibiting certain characteristics may require medical or surgical intervention.
The management approach for hemangiomas is dictated by the type and severity of complications. Treatment options can range from cutaneous laser therapy and pharmacotherapy with antiangiogenic agents to embolization and surgical excision. In rare cases, organ transplantation may be necessary [28]. Propranolol has emerged as a novel pharmacological agent for the treatment of infantile hemangiomas. Corticosteroids followed by chemotherapeutic agents, such as vincristine, are also employed [29].
Other vascular tumors
TA and KH are rare vasoproliferative tumors that manifest at birth or shortly thereafter [30,31]. Histologically, TA exhibits vascular tufts of densely packed capillaries arranged in a cannonball pattern, while KH consists of irregular nodules of compressed vessels with both vascular and lymphatic components [31]. The imaging features of TA and KH resemble those of other vasoproliferative neoplasms, but KH tumors are generally larger, less defined, more infiltrative, and associated with substantial flow voids due to the presence of numerous feeding and draining vessels [32]. Additionally, KH tumors are more likely to produce secondary destructive osseous changes. The treatment strategies for TA and KH are similar to those used for infantile hemangioma [33,34]. The presence of Kasabach-Merritt syndrome is a poor prognostic indicator for KH, with a mortality rate of 30%, thus necessitating aggressive treatment [35]. Kasabach-Merritt syndrome exhibits aggressive local behavior, and the absence of distant metastases makes extensive local excision and supportive care the fundamental pillars of therapy [34].
Angiosarcoma is a rare and aggressive neoplasm with a poor prognosis. It occurs in association with multiple infantile hemangioma, although it is a rare complication. The condition tends to affect females more than males, and the average age of onset is approximately 3.7 years. However, cases have been reported across all age groups [36].
VASCULAR MALFORMATIONS
Vascular malformations are abnormal growths of blood vessels that can manifest at birth or later in life [37]. Based on blood flow, they are primarily categorized into two types: slow- or low-flow malformations and fast- or high-flow malformations. Both genetic mutations and environmental factors contribute to the development of vascular malformations, which can cause a range of symptoms depending on their location and type. The treatment approach varies based on the severity and location of the malformation and may include medication, surgery, or embolization therapy [38].
Recent advancements in genetic and molecular research have revealed that most vascular malformations are associated with hyperactivated RAS/MAPK/ERK and PIK3/protein kinase B/mTOR pathways, which are triggered by inherited or somatic mutations [39]. Targeted molecular inhibitors that were originally designed to treat cancers with similar pathways, such as sirolimus and alpelisib, have demonstrated promising results in the treatment of vascular malformations [40]. The repurposing of cancer drugs has emerged as a central strategy in this field to improve patient outcomes and quality of life.
Low-flow malformations
Venous, lymphatic, and venolymphatic malformations are the most common types of vascular malformations, affecting up to 1% of the general population [41]. These malformations are characterized by abnormally formed and dilated veins or lymph vessels filled with serous fluid. Abnormal signaling pathways, such as the VEGF-C pathway, may be involved in the development of lymphatic malformations [42]. Purely venous or lymphatic structures are rare, with most of these malformations exhibiting both venous and lymphatic components [42].
Vascular malformations, which can be predominantly venous or lymphatic, may be present from birth or develop later in life. Predominantly lymphatic malformations are usually diagnosed before 2 years of age [28]. Venolymphatic malformations manifest as soft, easily compressible masses that may have a blue appearance and can increase in size with elevated venous pressure, such as during episodes of crying or Valsalva maneuvers [28]. Venous malformations also occur within bones; this distinguishes them from hemangiomas, which are not found in bone [43].
The management of venous malformations is predominantly determined by the size and location of the lesion. Although expectant management is suitable for some cases, others may necessitate intervention to alleviate pain, prevent potential organ damage, or avert airway obstruction. Treatment modalities range from palliative measures such as compression garments and close observation to more definitive procedures including sclerotherapy and surgical excision (Fig. 5) [44]. Nonetheless, due to the intricate and diffuse nature of these malformations, achieving complete and satisfactory treatment outcomes is challenging.
High-flow malformations
AVMs are typically present at birth, while arteriovenous fistulas are acquired malformations [28]. These conditions are characterized by a cluster of arterial and venous channels without a substantial solid mass. AVMs can manifest in various parts of the body, including the brain, bones, muscles, and subcutaneous fat. It is estimated that AVMs affect between 5 and 613 individuals per 100,000 population [45]. In some instances, AVMs may be linked to genetic disorders, such as hereditary hemorrhagic telangiectasia syndrome.
The first-line therapy for high-flow malformations, including AVMs and arteriovenous fistulas, is embolization. This is preceded by angiography to fully map the vessels. In certain cases, particularly in children, surgery or a combination of therapies may be required.
VASCULAR ANOMALIES IN THE HEAD AND NECK
Venous malformations
Over 40% of venous malformation cases occur in the head and neck region. Cervicofacial venous malformations typically grow in proportion to a child’s development and seldom regress [46]. These malformations are present at birth, but they may not be immediately noticeable. In particular, deeper lesions often do not become evident until later in childhood or adulthood [47].
Venous malformations are typically soft, compressible, and non-pulsatile, often presenting with a bluish discoloration. They have the potential to develop localized intravascular coagulopathy resulting from recurrent thromboses and blood stasis, as evidenced by elevated D-dimer concentrations [48]. Treatment modalities include percutaneous sclerotherapy, a procedure that involves the injection of a sclerosing agent under imaging guidance to induce damage and fibrosis in the affected vessels. Commonly used sclerosing agents include ethanol, sodium tetradecyl sulfate, and bleomycin, each of which is associated with a range of potential complications [49,50]. Surgery may be considered necessary based on the size and location of the lesion, as well as any impact on surrounding vital structures. The use of preoperative embolization with n-butyl cyanoacrylate glue can greatly reduce the risk of bleeding during the procedure and facilitate complete excision of the venous malformation [51]. Laser therapies, including neodymium-doped yttrium aluminum garnet and potassium-titanyl phosphate, may prove effective in reducing the need for resection and in debulking larger lesions. However, recurrence is a common issue in the management of venous malformations.
Capillary malformations
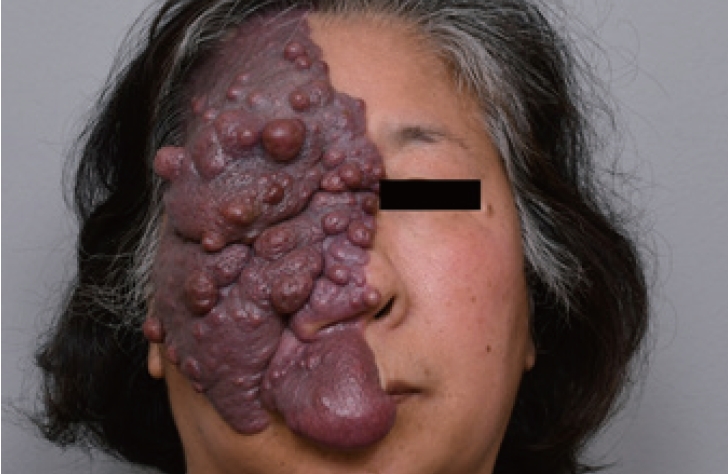
Capillary malformations (CMs) are the most common type of vascular malformation. They typically present as isolated cutaneous discoloration, such as port-wine stains and telangiectasia. The natural progression of CM involves a gradual darkening of the lesion, which is accompanied by epidermal thickening and increased nodularity [1]. Furthermore, some patients may develop soft tissue or bone hypertrophy in the affected areas. This can result in functional impairments affecting speech, swallowing, breathing, or vision, depending on the location of the malformation [52].
Numerous treatment options have been proposed for CM, but the primary method typically involves laser therapy. This specifically targets the vascular component to reduce the intensity of the discoloration. In cases involving an overgrowth of soft tissue, surgical intervention may be necessary to reduce excess tissue, restore symmetry, and enhance function. This is based on the established principles of facial and nasal aesthetic subunits. Such interventions may necessitate the use of local or expanded flaps.
Lymphatic malformations
Lymphatic malformations are benign abnormalities characterized by the improper development of lymphatic channels that fail to communicate with the normal lymphatic system. These are generally observed in regions rich in lymphatic vessels, such as the head and neck (45% to 52% of cases), mediastinum, axilla, retroperitoneum, and groin [53]. These lesions infiltrate the subcutaneous tissue and muscles, often situated above the mylohyoid muscles in the oral cavity, tongue, oropharynx, and salivary glands. Lymphatic malformations can lead to complications, including infection, cellulitis, and sepsis due to the leakage of superficial vesicles. They can also cause airway obstruction, necessitating a multi-staged and multidisciplinary approach. Other complications include dysphagia and internal hemorrhage, which can result in nerve compression [54]. The treatment strategy for lymphatic malformations depends on their location, size, and clinical presentation. Extensive and deep lesions are not suitable for surgery, making percutaneous sclerotherapy and medical treatment vital. Sirolimus treatment, either alone or in combination with sclerotherapy and surgery, may be effective for extensive and multi-spatial combined lymphatic malformations. Surgical resection is a common treatment option for lymphatic malformations that do not respond to sclerotherapy or other medical treatments. However, the malformation or scarring from previous surgery or sclerotherapy may distort normal anatomic relationships. Complete excision may not be necessary to achieve treatment goals, especially for larger and diffuse microcystic disease, as this can significantly increase morbidity. Children with lymphatic malformations affecting the head and neck, particularly larger trans-spatial lesions and those involving the tongue or floor of the mouth, may experience airway obstruction. This may require the insertion of a tracheostomy tube until the lesion is managed, resolving the obstruction and allowing for decannulation [55]. Medical treatment is becoming increasingly important in the management of extensive cervicofacial lymphatic malformations. Sirolimus has been found to be safe and effective in treating complex lymphatic malformations, reducing their size, pain, and associated infections.
Arteriovenous malformations
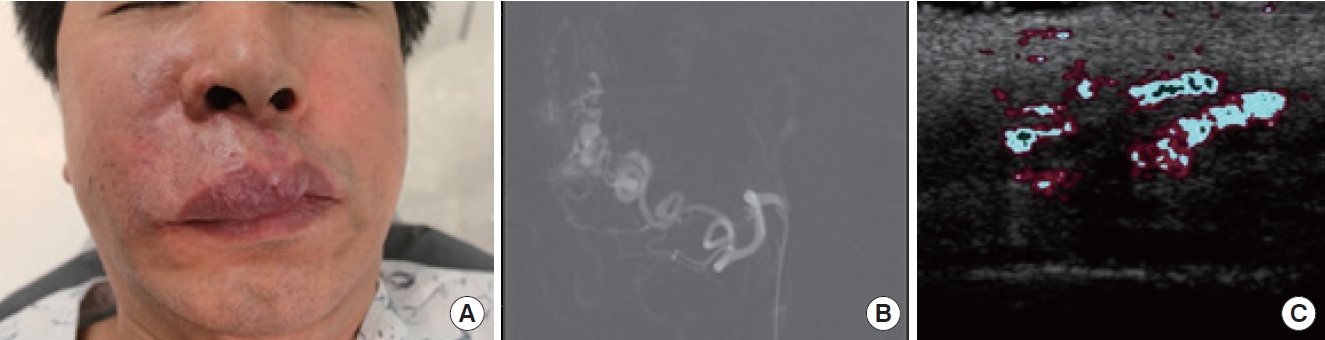
AVMs are congenital, but symptoms may not manifest until later in childhood or adolescence [56]. In some cases, AVMs may become rapidly enlarged due to factors such as infection, trauma, or hormonal changes during pregnancy [57]. Managing profuse bleeding from AVMs can be challenging, and high-output cardiac failure is a rare but possible complication.
The treatment of AVMs usually necessitates a team-based approach due to the complexity of these lesions. While curative treatment might be feasible for small, localized AVMs, the primary objective for more widespread lesions is symptom management and prevention of further progression. Endovascular embolization is frequently the first-line treatment, either alone or in combination with surgical resection. Surgical removal is typically performed within 48 hours of embolization to minimize blood loss and facilitate visualization of the lesion [58].
For AVMs in the head and neck region, surgical treatment involves removing the affected tissue, including the nidus, followed by immediate reconstruction of the area. Auricular AVM is a challenging condition that necessitates a collaborative and multi-departmental approach [59].
SYNDROMES PRIMARILY PRESENTING IN THE HEAD AND NECK
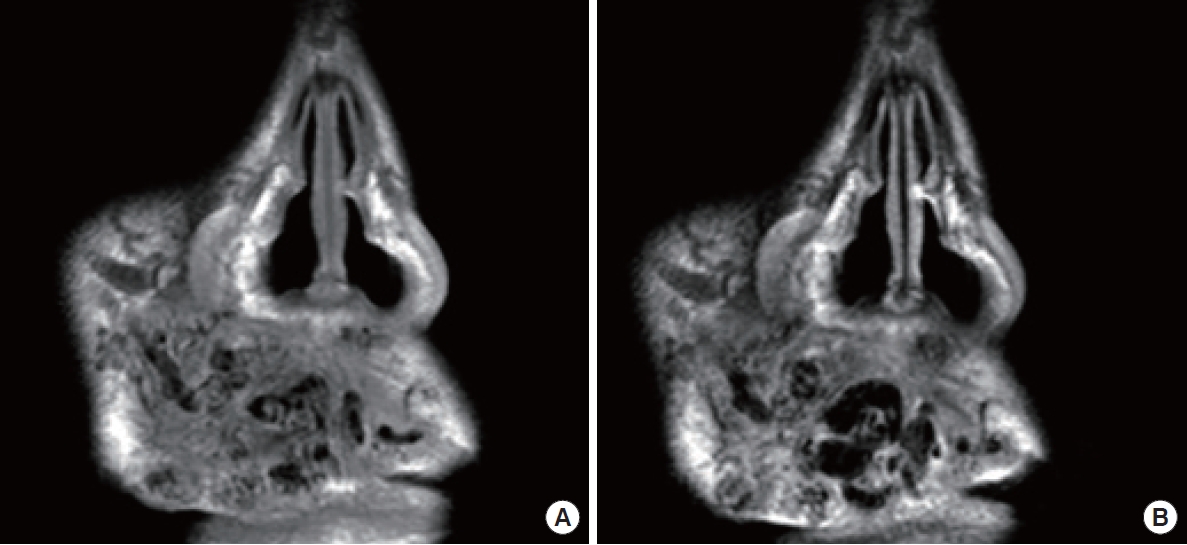
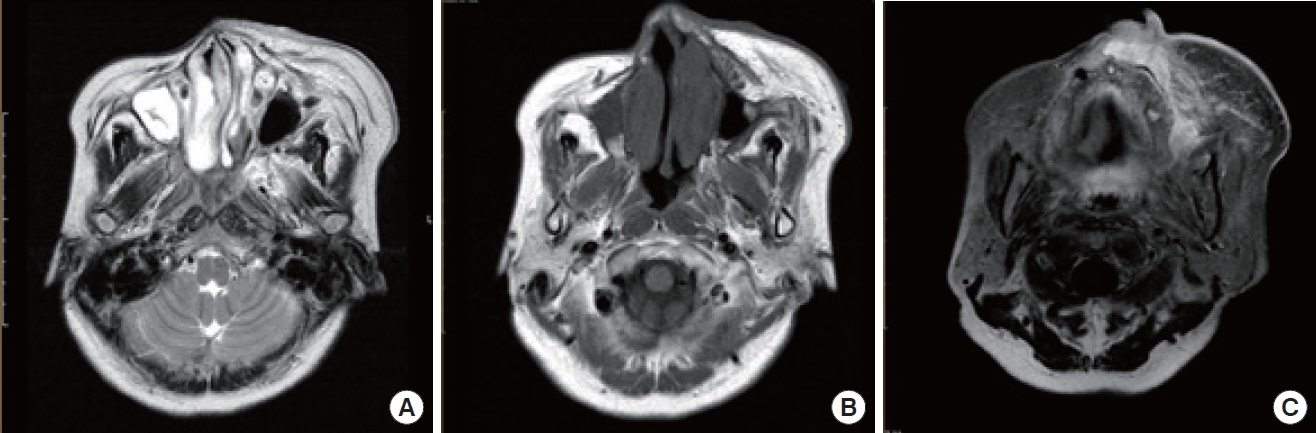
Syndromic vascular anomalies that can impact the face include Sturge-Weber syndrome (SWS) and Klippel-Trenaunay syndrome (KTS) [60,61]. SWS is an uncommon neurocutaneous disorder characterized by a port-wine stain on one side of the face and abnormal blood vessels in the brain (Fig. 6) [62]. This condition affects the first branch and potentially the second branch of the trigeminal nerve [63]. In addition, patients with SWS may experience seizures, developmental delays, and glaucoma [64]. However, these signs may not be apparent in younger children. In such cases, contrast-enhanced MRI can be used to detect abnormal pia mater enhancement on the same side as the facial CM.
In a substantial proportion of individuals with SWS (approximately 70%), ipsilateral choroidal vascular malformations are observed in tandem with facial CMs. This suggests a potential connection between these two clinical presentations. The prevalence of glaucoma in patients with SWS is approximately 30% [65]. The recent use of whole-exome sequencing has led to the discovery of a somatic activating mutation in the GNAQ gene. This gene encodes the guanine nucleotide-binding protein G(q) subunit alpha on chromosome 9q21 [66]. The mutation causes a change in the amino acid sequence of the Gaq subunit, which is integral to the G protein-coupled receptor signaling pathway [66].
In contrast, KTS is a rare vascular disorder characterized by a triad of symptoms: a port-wine stain, varicose veins, and overgrowth of soft tissue and/or bone [67]. While the port-wine stain typically appears on the leg, it may also present on the face. Individuals with KTS can exhibit lymphatic malformations and bleeding disorders [68,69]. These conditions are often unilateral, never crossing the midline, but can occur anywhere on the body. They are most commonly found on the face and in the cervical region [70]. KTS is a vascular disorder characterized by overgrowth, which is believed to stem from somatic mutations in the phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha gene [71].
Complications of KTS include deep vein thrombosis, pulmonary embolism, infections leading to sepsis, chronic coagulopathy, and symptoms such as bloody stools and epistaxis [72]. The differential diagnosis of KTS requires considering conditions such as Parkes Weber syndrome, cutis marmorata telangiectatica congenita, hemihyperplasia and multiple lipoma, Beckwith-Wiedemann syndrome, and Servelle-Martorell syndrome [4]. Recent research has highlighted a potential association between KTS and certain cancers, specifically lymphatic and bone cancer. As such, patients with KTS should receive regular monitoring for the development of these cancers, including Wilms tumor in children with KTS and basal cell cancer [73].
Overall, while both SWS and KTS may present with a port-wine stain, the clinical features and underlying pathophysiologies of these disorders are distinct. The predominant imaging feature of KTS is the excessive growth of subcutaneous soft tissues in the lower extremities [4].
Emerging evidence from retrospective studies indicates that a combination of low-dose aspirin and vitamin D may be a promising treatment option for SWS [62]. Additionally, prospective clinical trials have established the effectiveness of cannabidiol and sirolimus. The pre-symptomatic administration of low-dose aspirin and antiepileptic drugs has shown positive results in delaying the onset of seizures in some patients [62].
For the management of KTS, a conservative approach is recommended, utilizing non-invasive measures for symptomatic treatment. Elastic or non-elastic compression stockings, supplemented with psychological support, are the most effective treatments for the majority of cases [74]. Anticoagulants can be administered prophylactically prior to surgery or in instances of acute thrombosis [67]. While conventional sclerotherapy may be effective for smaller malformations, it is not suitable for treating larger ones [75]. Pain is a common symptom of KTS, affecting up to 88% of those with the condition, and requires frequent management [76]. Surgical intervention is generally reserved for symptomatic cases, with laser treatment frequently used for port-wine stains [77].
OTHER SYNDROMES PRESENTING WITH VASCULAR ANOMALIES
Blue rubber bleb nevus syndrome (BRBNS) is a rare condition characterized by multiple blue-to-black, rubbery skin nevi as well as venous malformations in the gastrointestinal tract [78,79]. Common symptoms include gastrointestinal bleeding, anemia, and coagulation disorders. Diagnostic tools such as barium contrast studies and CT scans are useful in diagnosing the syndrome [79,80]. Recent studies suggest a potential connection between BRBNS and mutations in the TEK gene, which influences angiopoietin-TIE2 signaling [81]. While no curative therapy is available, treatment options for BRBNS-associated gastrointestinal bleeding include endoscopic sclerotherapy, embolization, and surgical resection. Sirolimus therapy has shown promise in treating complicated cases [82].
Proteus syndrome is a rare congenital disorder characterized by soft tissue swelling, connective tissue nevi, vascular malformations, and asymmetric bone overgrowth [83]. It was initially thought to be associated with PTEN mutations but was later linked to Akt-1 kinase mutations [84]. The use of miransertib as targeted therapy has demonstrated promising results in reducing overgrowth and improving quality of life in patients with Proteus syndrome.
Maffucci syndrome is a rare genetic disorder characterized by vascular malformations, enchondromatosis, and an increased risk of malignant tumors [85]. Imaging findings include phleboliths and bone lesions. Due to the increased risk of malignancy, long-term monitoring is essential [86]. While sirolimus appears to be a promising treatment, additional research on this option is required [87].
Parkes Weber syndrome is frequently confused with KTS. The distinguishing factor is the presence of high-flow malformations [88]. This condition is triggered by mutations in the RASA1 gene [89]. Treatments for high-flow malformations include embolization, surgical resection, and stent-graft implantation.
PHACE syndrome is an uncommon condition characterized by various clinical and imaging findings, including posterior fossa malformations, hemangiomas, arterial anomalies, cardiac defects, eye or endocrine anomalies, and sternal defects [90]. Treatment depends on the specific manifestations of the syndrome.
Kasabach-Merritt syndrome is a pediatric condition characterized by vascular tumors, decreased platelets, anemia, and coagulation abnormalities [91]. Treatment options include extirpation, steroid therapy, vincristine, interferon, radiation therapy, and sirolimus. Differentiation between Kasabach-Merritt syndrome and Kasabach-Merritt–like phenomenon is crucial for appropriate treatment [92].
RECONSTRUCTIVE ISSUES AMONG PATIENTS WITH VASCULAR ANOMALIES
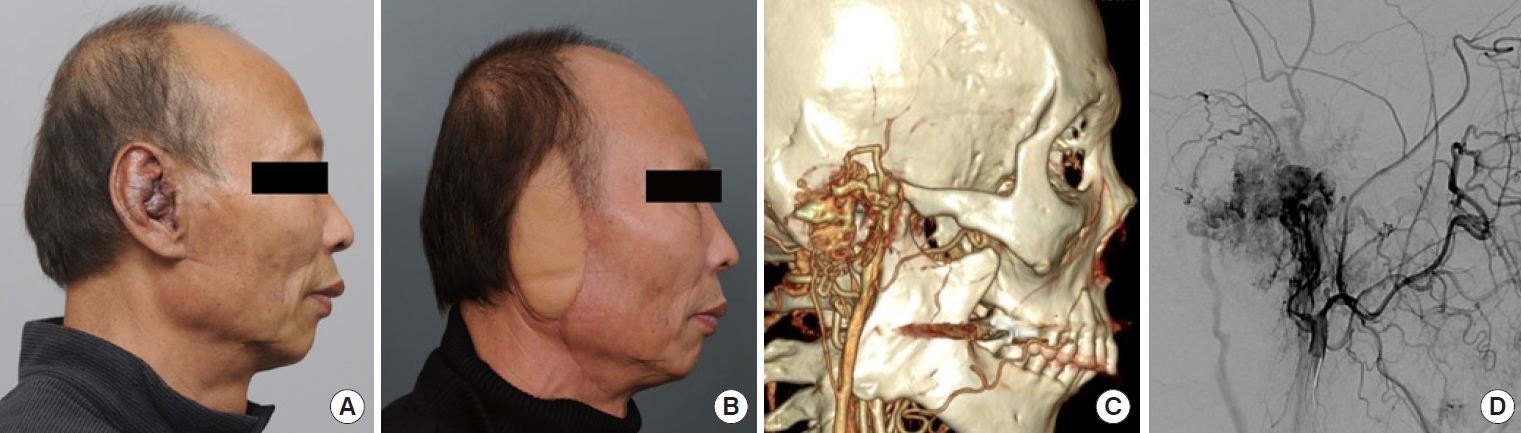
Challenges associated with reconstructing vascular anomalies may involve the removal of the affected tissue, restoration of normal blood flow, and improvement of cosmetic appearance, among other considerations. Treatment approaches depend on the specific type and location of the anomaly, as well as the individual needs of the patient (Fig. 7).
Facial vascular anomalies can be particularly challenging because they affect the appearance and function of the facial features. Depending on the type and severity of the anomaly, reconstructive surgery may be necessary to address the issue. For instance, if the vascular anomaly presents as a hemangioma or a port-wine stain, laser therapy or surgical excision may be considered to improve the cosmetic appearance of the affected area. If the anomaly is an AVM or lymphatic malformation, surgical removal or embolization may be necessary to prevent complications such as bleeding, swelling, or infection.
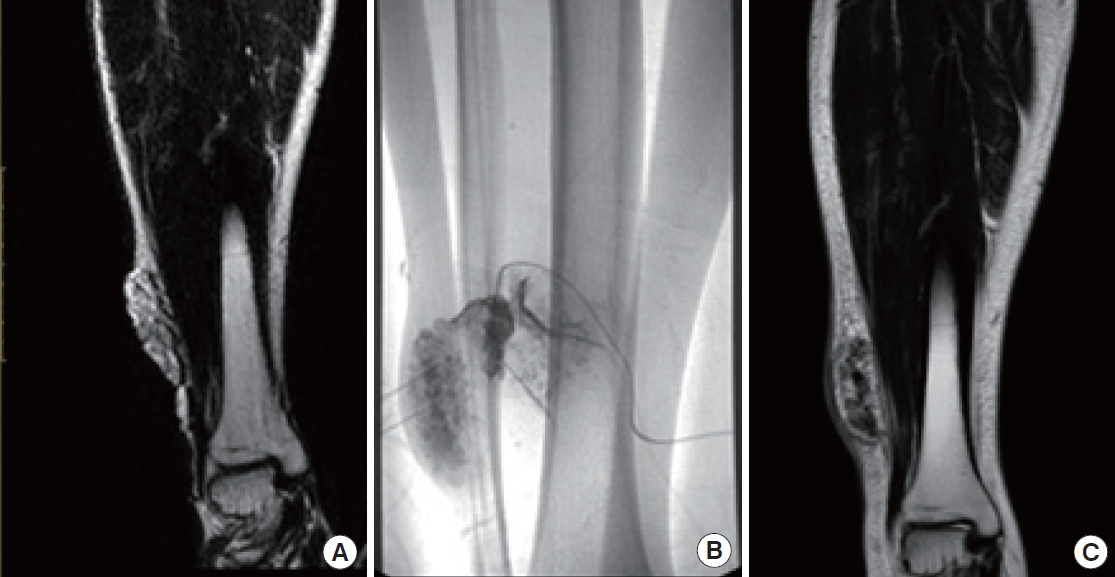
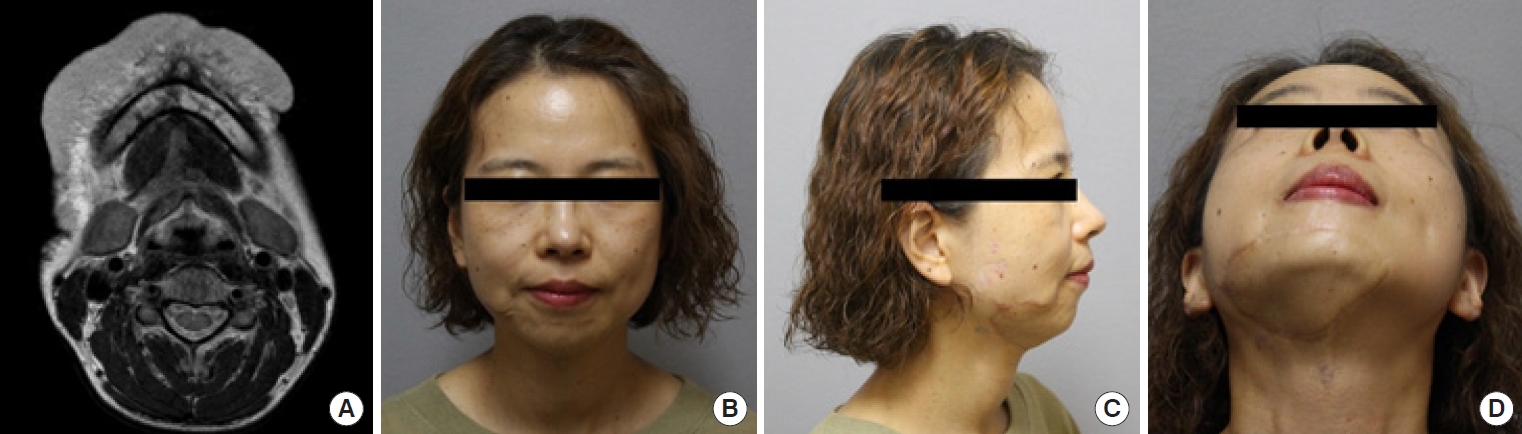
Complete surgical resection is the optimal treatment for vascular anomalies, but it may not be feasible when the anomaly is extensive or symptomatic. Radical excision should not be attempted without considering the potential for postoperative functional loss or impairment (Fig. 8). For larger lesions, a combination therapy approach involving preoperative angiography is recommended. This involves preoperative angiography with selective embolization, followed by definitive resection within 1 to 14 days [93,94]. According to a study by Tark and Chung [94], using a free radial forearm flap for reconstruction after resection can result in the histological regression of residual lesions to normal tissue over time. Furthermore, the use of an anterolateral thigh flap underscores the effectiveness of employing well-vascularized tissue coverage after resection, particularly when residual malformations remain after the removal of an extensive lesion (Fig. 9) [95]. For thin-skinned areas such as the chin, where good aesthetic outcomes are possible, superficial circumflex iliac artery perforator flaps should be considered as a viable option (Fig. 10).
SUMMARY
The ISSVA has established a widely recognized classification system for vascular anomalies. In recent years, notable advancements in genetic testing and imaging techniques have improved the diagnosis of these conditions. Genetic testing has become increasingly sophisticated, facilitating the identification of specific genetic mutations that inform treatment decisions. Furthermore, imaging techniques such as MRI and CT have greatly improved our ability to visualize and detect vascular abnormalities, enabling more accurate diagnoses. Despite these recent advancements in genetic testing and imaging techniques, diagnosing vascular anomalies can be challenging. This is due to the rarity of these conditions and the broad spectrum of potential symptoms. When considering reconstructive surgery for facial vascular anomalies, it is important to consider both the functional and cosmetic outcomes of the procedure. In some cases, surgery may improve the cosmetic appearance of the face but also lead to functional deficits, such as loss of sensation or movement. Therefore, a comprehensive multidisciplinary approach involving specialists from dermatology, radiology, and genetics is often required to ensure accurate diagnosis and effective management of these conditions. Overall, the treatment approach for facial vascular anomalies depends on the type, size, location, and severity of the anomaly, as well as the individual needs and goals of the patient. A thorough evaluation by a team of specialists can determine the most appropriate and effective treatment plan.