Current concepts of craniofacial fibrous dysplasia: pathophysiology and treatment
Article information

Abstract
Fibrous dysplasia is an uncommon genetic disorder in which bone is replaced by immature bone and fibrous tissue, manifesting as slow-growing lesions. Sporadic post-zygotic activating mutations in GNAS gene result in dysregulated GαS-protein signaling and elevation of cyclic adenosine monophosphate in affected tissues. This condition has a broad clinical spectrum, ranging from insignificant solitary lesions to severe disease. The craniofacial area is the most common site of fibrous dysplasia, and nine out of 10 patients with fibrous dysplasia affecting the craniofacial bones present before the age of 5. Surgery is the mainstay of treatment, but the technique varies according to the location and severity of the lesion and associated symptoms. The timing and indications of surgery should be carefully chosen with multidisciplinary consultations and a patient-specific approach.
INTRODUCTION
Fibrous dysplasia (FD) is a nonheritable genetic disorder, in which normal bone is replaced by immature, haphazardly distributed fibro-osseous tissue, resulting in deformity, fractures, pain, and functional impairment. Due to varying degrees of mosaicism, its clinical spectrum can range from asymptomatic, solitary lesions (monostotic) to severely disabling multiple skeletal lesions (polyostotic) and extra-skeletal disease, such as hyperpigmented skin lesions and hyperfunctioning endocrinopathies.
EPIDEMIOLOGY AND CLASSIFICATION
FD accounts for 5% to 7% of all benign bone tumors. It is described as monostotic if a single bone is involved, and polyostotic if multiple bones are involved. The monostotic form is reported to occur in 70% to 80% of all FD patients, and the most common site of involvement is the femur [1]. Of patients with FD affecting a craniofacial bone, 56% had monostotic disease, 47% had polyostotic disease and 7% had McCune-Albright syndrome (MAS) [2]. Since multiple bones are interconnected nearby in the craniofacial and gnathic areas, as long as there is only one disease focus, the lesion is considered monostotic by most authors. However, some authors consider the extensive involvement of multiple craniofacial bones as polyostotic disease. The most commonly involved craniofacial bone is the maxilla (or zygomaticomaxillary complex), followed by the mandible, frontal, sphenoidal, ethmoidal, parietal, temporal, and occipital bones. Craniofacial involvement in monostotic FD has been reported in 10% to 25% of cases, but craniofacial involvement was reported to be present in 50% to 90% of cases of the polyostotic form [3,4].
The polyostotic form accounts for 20% to 30% of all patients, involves several bones with multiple foci, and more commonly presents on only one side of the body, but can be found bilaterally [5]. Historically, symmetric and extensive polyostotic FD involvement of the craniofacial bones led to changes in patients’ face that were considered to resemble a “lion’s face,” for which reason it was called “leontiasis ossea” [6]. In the spectrum of polyostotic FD, various associated disorders are known and named as different syndromes. If polyostotic FD presents with only pigmented café-au-lait skin lesions, it is called Jaffe-Lichtenstein syndrome. Polyostotic FD with skin lesions and hyperfunctioning endocrinopathies is called MAS, and it accounts for 3% of all FD. If intramuscular myxomas are present with polyostotic FD, it is called Mazabraud syndrome (Fig. 1) [7].
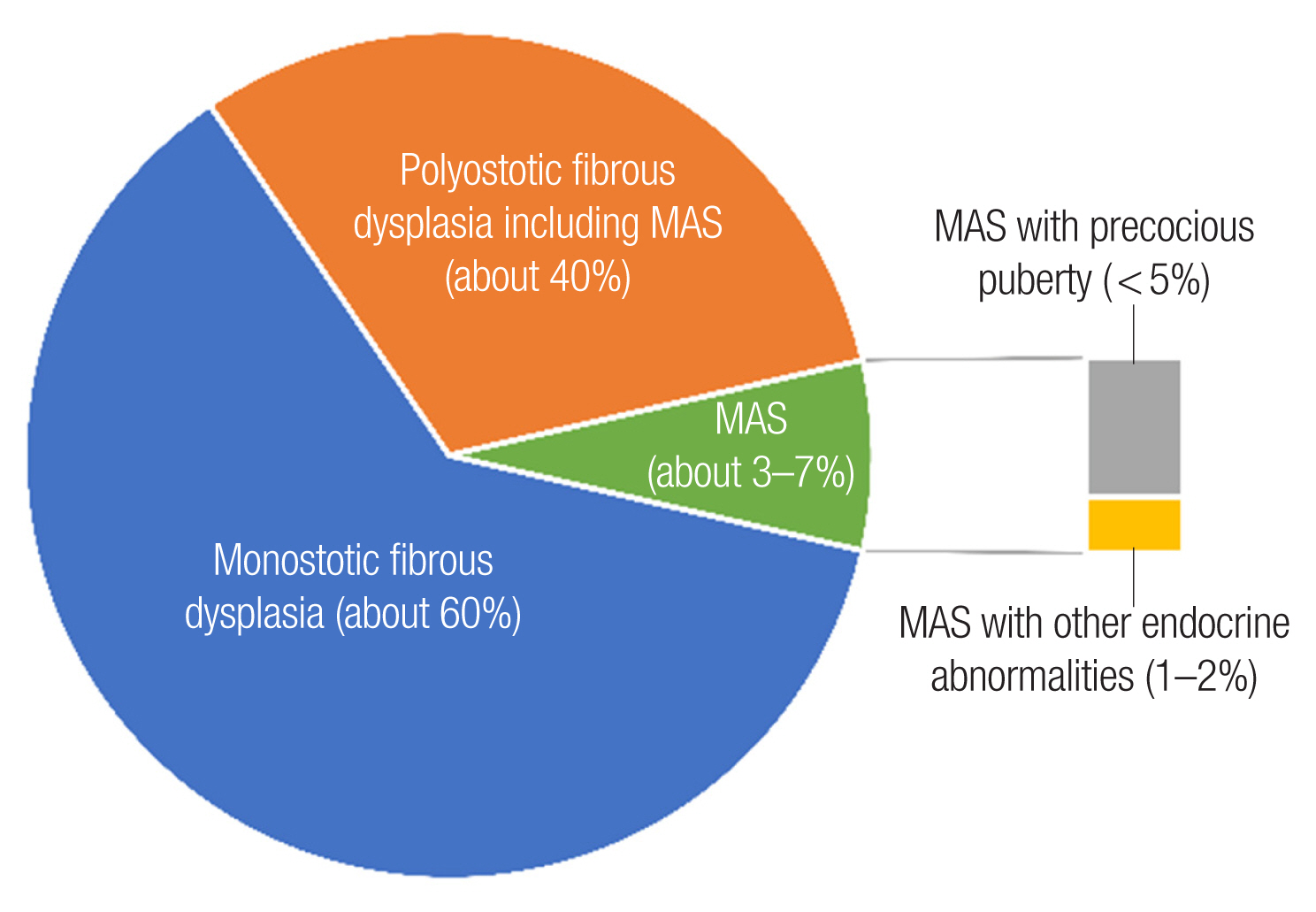
Distribution of fibrous dysplasia according to its classification. MAS, McCune-Albright syndrome.
Since mild, asymptomatic FD is found incidentally on imaging studies, many patients may be undiagnosed. Furthermore, FD lesions are poorly characterized, and there is a possibility that monostotic FD is overdiagnosed for solitary bone lesions. Not all patients diagnosed with monostotic FD undergo whole-body and endocrine studies; thus, the true prevalence of polyostotic FD and its many subtypes is difficult to know.
PATHOGENESIS AND PATHOPHYSIOLOGY
FD is caused by sporadic post-zygotic mutations in the GNAS gene, which is located on chromosome 20 and encodes the α-subunit of the Gs stimulatory protein [5,8]. Mutations occur at one of two positions: Arg201 (>95% of reported cases) or Gln227 (<5%) [9,10]. The mutation disrupts the intrinsic GTPase activity of Gsα, resulting in constitutive receptor activation and inappropriate cyclic adenosine monophosphate (cAMP)-mediated signaling, followed by excess formation of cAMP in mutated cells (Fig. 2). Since the skin, bone, and endocrine systems are derived from the ectoderm, mesoderm, and endoderm, respectively, the timing of the mutation is associated with the severity and extent of the disease. Therefore, in MAS, the mutation must occur early in embryogenesis, prior to the derivation of the three germ layers [11]. In contrast, the less severe monostotic FD phenotype is assumed to be caused by a mutation event occurring later in development. This hypothesis also provides a biological basis for the observation that monostotic FD does not progress to polyostotic disease over time [12]. The elevated cAMP results in the formation of bone marrow stromal cells (BMSCs) that are unable to differentiate into normal marrow components, including osteoblasts, adipocytes, and hematopoiesis-supporting cells [13,14]. Impaired differentiation of osteogenic progenitors into mature osteoblasts and osteocytes causes the proliferation of immature osteoprogenitors and produces excess amounts of abnormal bone matrix, consisting predominantly of woven bone. Overexpression of receptor activator of nuclear factor kappa-B ligand and interleukin-6 by mutated osteoblastic cells activates osteoclasts; in turn, bone resorption is increased, and bone is replaced with overproduced disorganized collagenous matrix so that the FD lesion expands [14,15]. There are three distinctive patterns in histology: the traditional Chinese character form, the Pagetoid type, and the hypercellular type. The common part is that the normal bone is replaced with cellular fibrous tissue containing various inorganic substances. Fibrosis of the cranial facial bone shows different histological patterns from lesions occurring in the torso. First, it is difficult to observe cartilage differentiation unless there is a history of fractures (pathological fractures are also rare in the maxillary bone compared to the iliac bone). Second, compared to the Chinese character type, which is mostly composed of woven bones, the Pagetoid type and the hypercellular type have considerable bone tissue and forms a layered structure to some extent. Third, some of the osteoblastic rimming surrounding the bone marrow can be observed (Fig. 3). These histological findings themselves do not predict the prognosis of the disease, but the possibility of delayed diagnosis or errors by pathologists should also be kept in mind if imaging and clinical findings are not properly considered together. Dysplastic bone undergoes active remodeling, and nearly 50% of patients with FD/MAS exhibit some degree of renal phosphate wasting [16].

Pathophysiology of fibrous dysplasia and current treatment targets. BMSC, bone marrow stromal cell; IL-6, interleukin-6; PDGF-B, platelet-derived growth factor subunit B; FGF-23, fibroblast growth factor-23; NFG, nerve growth factor; GTP, guanosine triphosphate; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; CREB-P, cAMP response element-binding protein; CREM-P, cAMP response element modulator protein; AP-1, activator protein 1.
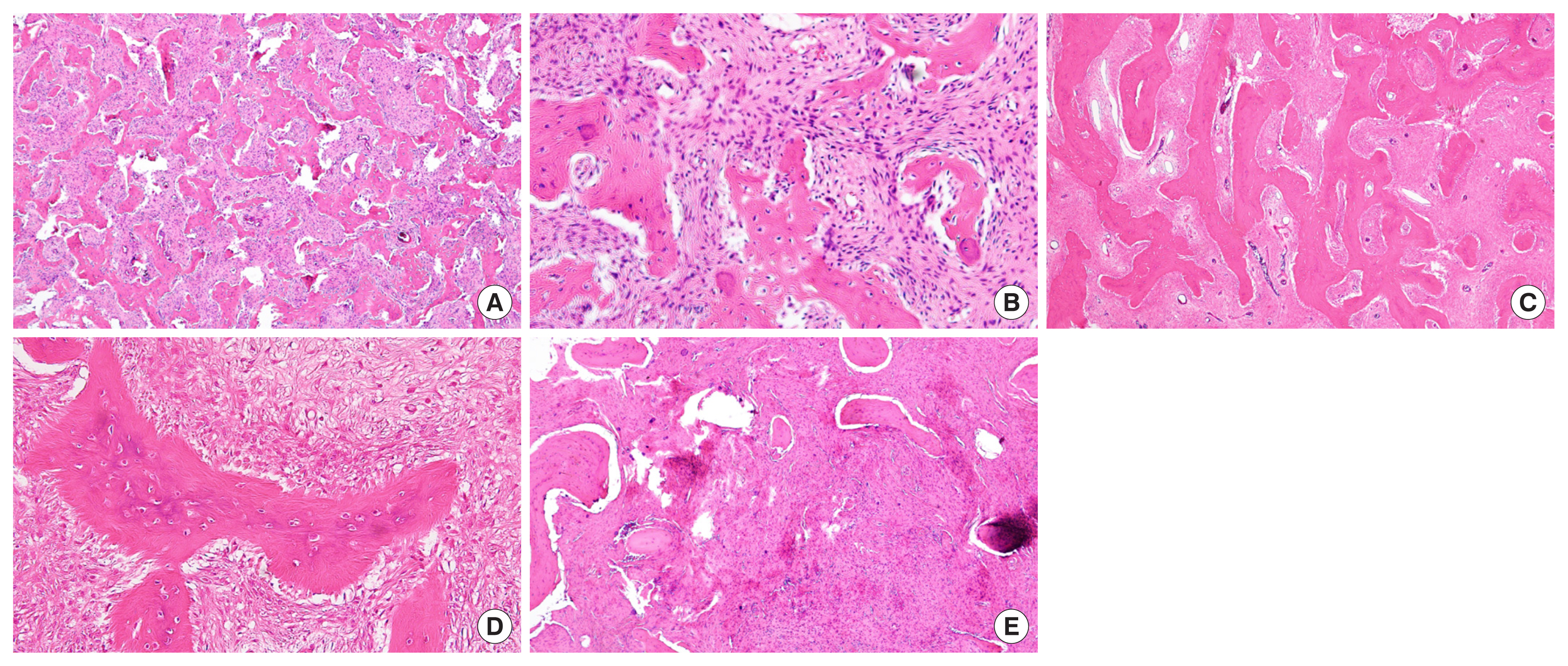
Histologic features of craniofacial fibrous dysplasia (all images are in hematoxylin and eosin stain). (A) Typical low-power appearance. Irregular, curvilinear, and immature bony trabeculae surrounded by fibrous background (zygoma, ×40). (B) Osteoclast activity and focal osteoblastic rimming is observed at high-power field (zygoma, ×200). (C) Relatively preserved continuity and lamination of bony trabecula. (skull, ×40) (D) Absent of osteoblastic rimming (skull, ×200) (E) Hypercellular lesion with abundant fibrous tissue (mandible, ×100).
CLINICAL PRESENTATION AND NATURAL HISTORY
The clinical presentation of FD can be at any age, although it mostly occurs before 15 years of age and usually takes place around 10 years of age. FD in the craniofacial area develops particularly early, with the majority of the lesions established by 3 years of age [17]. FD lesions typically manifest during the first few years of life, and expand during childhood and adolescence. Clinically significant bone lesions are usually apparent by the age of 5 years, with almost no significant lesions appearing after age 15 [17]. A tendency for the disease to progress throughout adolescence and stop its progression after adolescence may or may not be observed. FD lesions may become less active in adulthood, which might be related to apoptosis of mutation-bearing BMSCs [18]. However, there are also several reported cases of FD remaining or becoming active well into adulthood [4].
Monostotic lesions tend to enlarge in proportion to skeletal growth and the prognosis is generally good. Polyostotic disease has impacts proportional to the extent of disease and by adolescence, many patients with widespread polyostotic FD and MAS have severe deformities and functional impairment.
Skeletal manifestations
Unlike the bones of the extremities, which typically present with fractures, craniofacial FD typically shows slow-growing lesions causing gradual painless swelling and facial asymmetry. Due to the high concentration of vital structures in this area, these lesions can result in serious complications and present unique challenges to management. Besides cosmetic deformities, other symptoms may arise related to the lesions located in close proximity to vital structures. Furthermore, since a significant proportion of craniofacial FD patients may experience bone pain, it is important to rule out odontogenic pathology [19]. Dental problems such as malocclusion, orbital dystopia, visual disturbance, hearing impairment, nasal congestion, and obstruction may occur. However, a series of 66 patients diagnosed with craniofacial FD found that 36% were asymptomatic [7].
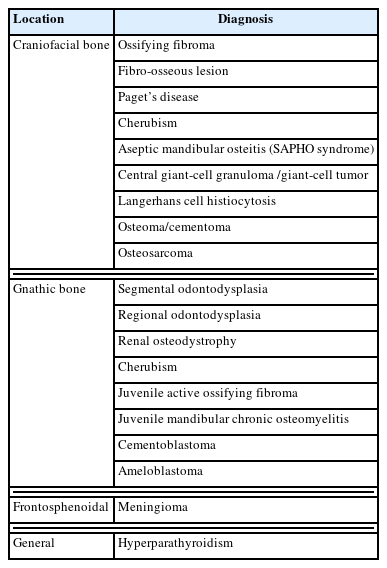
If a rapidly growing lesion or lesion with pain or paresthesia is noted, other diagnoses, such as aneurysmal bone cyst or growth hormone (GH) excess, should be considered and a biopsy is recommended. The risk of bone biopsy should be considered as well as its benefits, because in most cases, the clinical history, physical examination, and radiographic findings are sufficient for high-probability diagnosis. Furthermore, the differential diagnosis of FD is not possible solely on the basis of a pathologic evaluation; instead, the age of presentation, clinical extent, and severity are more important factors (Table 1) [4,9,20,21].
Craniofacial FD lesions are some of the earliest to be detected, but can remain “silent” until deformity or growth occurs. A study of 109 patients over 32 years at the National Institutes of Health found that 90% of craniofacial lesions were present by 3.4 years. In the extremities, 90% of lesions were present by 13.7 years, and, in the axial skeleton, 90% of lesions were present by 15.5 years [17].
Plain radiography, computed tomography (CT), magnetic resonance imaging (MRI), and bone scintigraphy are all modalities that have been used in the evaluation of FD. In the craniofacial area, non-enhanced CT is most commonly used and constitutes the best imaging modality for diagnosis and follow-up assessments. In general, the radiographic appearance can be variable because it is influenced by the proportion of mineralized tissue and fibrous tissue in the lesion. This variability results in a range of appearances, from the classically described ground-glass appearance with ill-defined borders, to a mixed radiolucent-opaque lesion, to one that is primarily radiolucent. The radiographic appearance of FD also varies by both age and location. The radiographic appearance of FD changes from a homogeneous ground-glass appearance during childhood and adolescence to a less radiolucent, more mixed and heterogeneous appearance thereafter, and older patients, and those treated with bisphosphonates have been noted to have radiographically sclerotic lesions [22]. Craniofacial FD lesions are not well-demarcated, often crossing bony sutures, but not breaching the bony cortices. A CT scan can help to delineate the specific nature of an FD lesion and its surrounding structures. One of the most characteristic radiographic features of FD is the involvement of the sphenoid bones and the skull base. In cases of sphenoid involvement, the orbital canals should be evaluated for narrowing. Baseline and periodic CT scans of the head should be performed in children, usually every 2 years or less frequently based on the localization and severity of the lesion(s). Regular imaging is not indicated in adults, and the timing of the scans should be based on symptoms, at most every 5 years in those without symptoms.
Extra-skeletal manifestations
Café-au-lait spots are generally the earliest manifestation of MAS, presenting at or shortly after birth. In FD, characteristic jagged “coast of Maine” borders are observed, in contrast to the smooth “coast of California” borders in neurofibromatosis, and these spots tend to be distributed along the midline of the body, reflecting patterns of embryonic cell migration (Fig. 4) [23].

Presentation of café-au-lait spots. (A) Café-au-lait spots in fibrous dysplasia show more complex margins. (B) Café-au-lait spots in neurofibromatosis tend to have smoother borders.
Endocrinopathies typically become apparent during childhood and persist into adulthood. Precocious puberty is the most common presenting symptom in girls, affecting approximately 85% of patients with MAS [23,24]. In contract, testicular abnormalities affect approximately 85% of boys and men, and may present with macroorchidism on an examination. Hyperthyroidism, GH excess, and neonatal hypercortisolism may occur. GH excess is an important contributor to morbidity in patients with craniofacial FD, and is associated with a higher prevalence of macrocephaly, optic neuropathy, and hearing loss [25].
Certain cancers have been reported in association with FD/MAS, including osteosarcoma, thyroid cancer, and breast and testicular cancers [26]. If a rapidly expanding lesion is associated with new localized pain or paresthesia, malignancy and aneurysmal bone cysts should be suspected, and MRI is recommended modality of diagnosis. Aneurysmal bone cysts are benign, rapidly expanding cystic lesions that can cause severe pain and compression sequelae and are best treated with surgery [27].
MANAGEMENT
The goal of the management of FD is to optimize function and minimize morbidity, not to achieve a complete cure or cessation of disease progression. The treatment strategy may vary according to disease burden. Treatment often consists of surgery to preserve function and reduce pain, as well as conservative techniques that may include a combination of physical therapy, orthoses, avoidance of prolonged immobilization, and management of underlying endocrinopathies [28]. Thus, once the diagnosis is made, a team approach with interdisciplinary consultation for identifying and managing extra-skeletal disease is essential.
Medical therapy
Currently, no medical treatment has proven efficacy in treating, curing, or definitively stopping the progression of FD. Pain control is the mainstay of treatment. About 40% of patients with craniofacial FD present with pain, and bisphosphonates are often used [21,29,30]. Bisphosphonates are anti-resorptive agents that decrease bone turnover by inhibiting osteoclasts. However, studies of the effects of bisphosphonates have shown conflicting results. The only randomized, double-blind placebo-controlled trial of a bisphosphonate in FD showed that oral alendronate had no effect on bone pain or the radiographic appearance compared to placebo [30]. Early reports on intravenous pamidronate showed beneficial effects on bone pain, but inconsistent effects on the radiographic appearance of FD [31–33]. Concerningly, recent evidence has established that patients with FD are at risk for osteonecrosis of the jaw as a consequence of bisphosphonate therapy [34]. Therefore, intravenous bisphosphonates are a treatment option for FD-related bone pain, but caution is needed for long-term treatment with potent intravenous formulations [34]. The proposed protocols of intravenous pamidronate or zoledronate differ from those published in articles. However, generally a total of 180 mg of pamidronate is given at 60 mg/day for 3 days or 90 mg/day for 2 days every 6 months for 12 or 18 months (Table 2) [21,29]. Few medical targets other than bisphosphonate exists and clinical trials are ongoing for the treatment and management of FD (Fig. 2) [35,36].
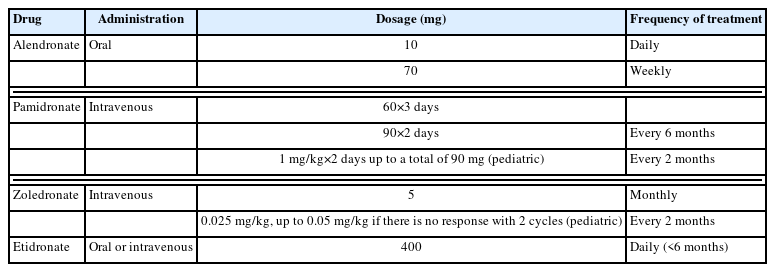
Regimens of commonly used bisphosphonates in fibrous dysplasia
With the use of an intravenous bisphosphonate, clinicians should consider using calcitriol with calcium supplements for 4 days after the first injection to maintain calcium homeostasis, especially in those with a high disease burden. The treatment cycle should be ceased if there is no response after two to three cycles.
Surgical management
Although the primary aim of treatment should always be to preserve function, improvement of aesthetics by treatment of the primary deformity is also a meaningful goal. The control of secondary diseases, such as mucocele, aneurysmal bone cysts, and some sarcomas, is also important. Regular monitoring for functional deficits, including clinical examination for sensory, motor, ophthalmologic and audiologic functions at least annually, photographs, and periodic facial CT imaging are mandatory for patients with craniofacial FD [37]. Bony lesions can be categorized as quiescent (stable, no growth), non-aggressive (slow growth), or aggressive (rapid growth with or without pain, paresthesia) [22]. Observation with close monitoring can be a treatment option for some period of time in cases of stable disease. To minimize recurrence, elective surgery should be postponed until after skeletal maturity, when FD lesions tend to become more quiescent. However, cases of aggressive disease with significant growth potential, particularly in the setting of endocrinopathies and GH excess, are surgical candidates, for which complete excision is desirable in patients of any age at presentation.
Advanced imaging techniques and three-dimensional analyses of scans, together with virtual surgical planning for subtotal excision or complete excision of the lesion and reconstruction with autologous tissue or implants made by computer-aided manufacturing and patient-specific design, should be regarded as the standard of care in surgery for FD of the craniofacial skeleton [38,39]. Simple curettage is not recommended as a primary option, as it is ineffective and may increase the risk of complications [40–42]. Other options for surgical intervention are burring the lesion to reduce bulk and achieve symmetry. However, while procedures involving resection with reconstruction have greater morbidity, they show a lower rate of recurrence than recontouring procedures (45% vs. 82%, respectively) [43].
Recontouring procedures, such as burring or shaving, are performed quite commonly and are beneficial in some anatomical regions [44]. These procedures can also be supported by surgical navigation systems, which may assist in overcoming some difficulties of achieving symmetry and avoiding damage to critical anatomic structures. However, the outcomes are often suboptimal due to frequent postoperative regrowth of lesions (in up to 68% of cases), especially in the setting of untreated GH excess [45,46]. Thus, GH excess and other endocrinopathies should be screened for and treated prior to surgery. Untreated GH excess is associated with vision loss; therefore, GH excess should be treated as early as possible (Fig. 5) [22].
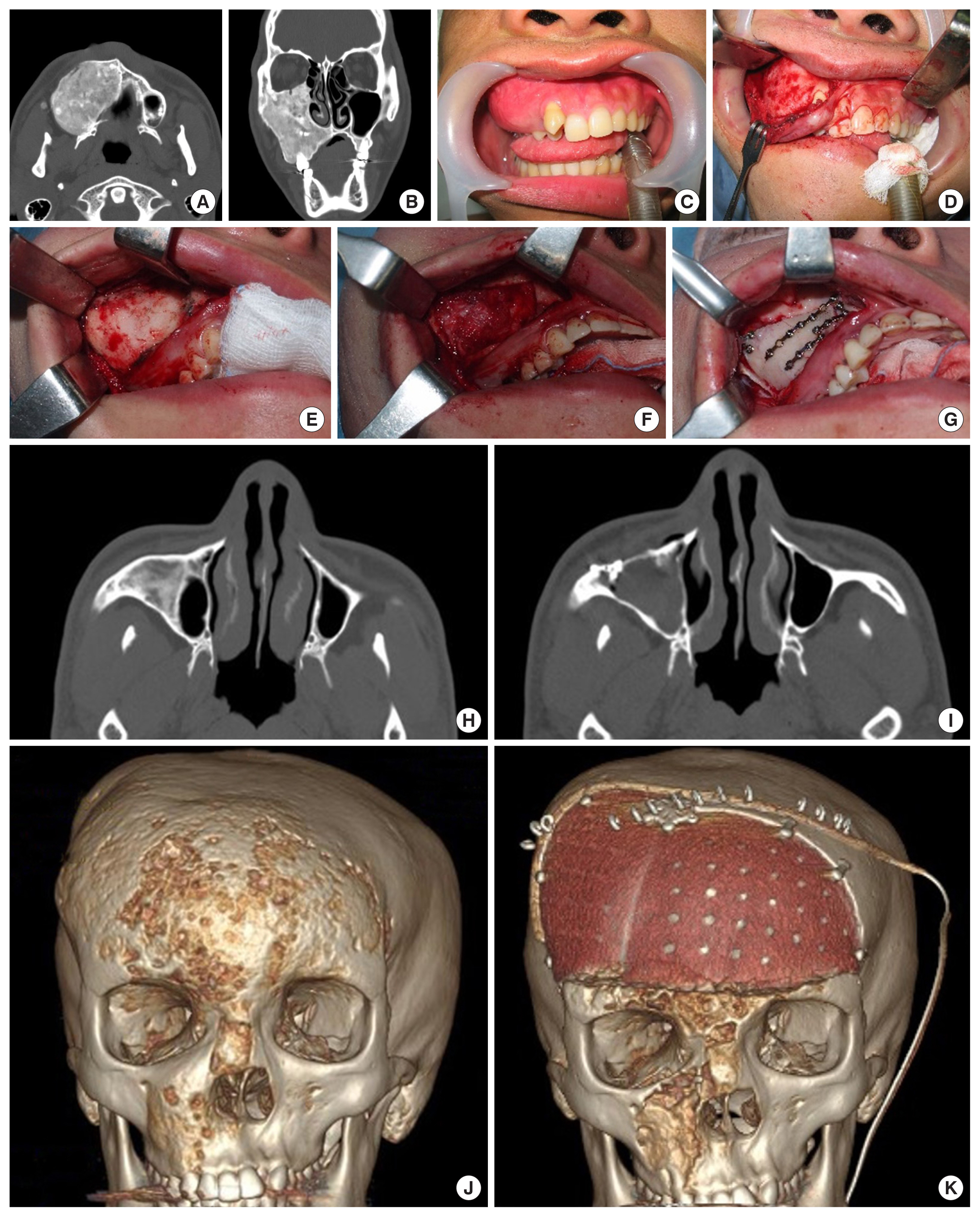
Surgical management of craniofacial fibrous dysplasia. (A-D) Simple shaving of the maxillary alveolar bone lesion with preservation of normal dentition. (E-I) Decortication with a maxillary anterior wall bone graft. (J, K) Radical excision of a frontal bone lesion and reconstruction with a polyetheretherketone patient-specific implant was done, while the periorbital and maxilla areas were recontoured with shaving.
The surgical approach may vary depending on tumor location (Table 3, Fig. 6) [40,47].

Treatment principles of craniofacial fibrous dysplasia
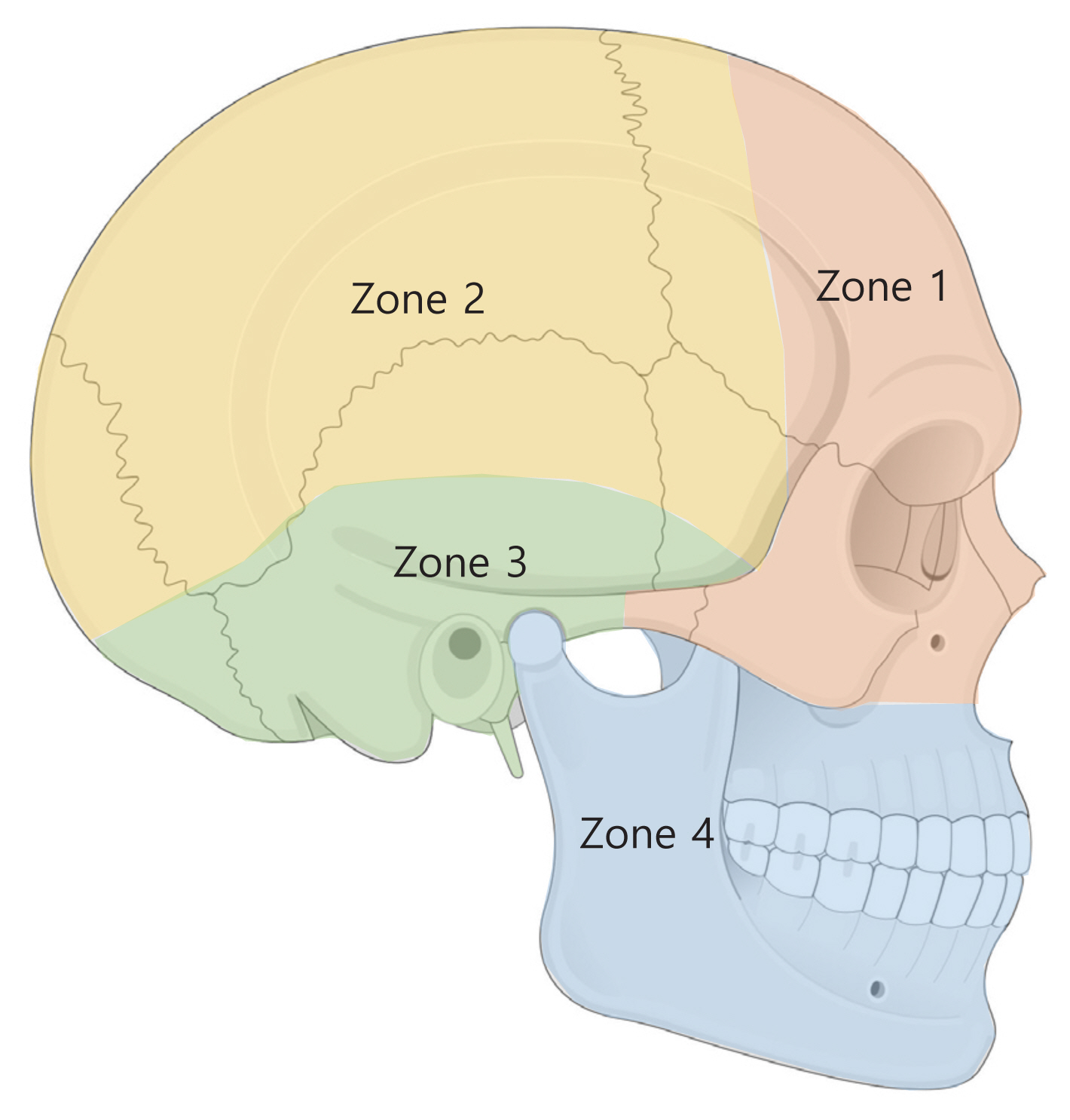
In zone 1 (fronto-orbital, zygomatic, and upper maxillary regions), complete excision with reconstruction is advisable since this is the most aesthetically apparent area. A bicoronal incision is used in frontal bone lesions. After osteotomies, for complete resection, split calvarial grafts can be assembled into a three-dimensional construct or titanium mesh, or a patient-specific implant in various materials can be used [48,49]. Lesions involving the supraorbital and infraorbital rims may be treated less aggressively, with the use of a CAD-assisted approach (i.e., intraoperative navigation and a three-dimensional constructed patient-specific implant) [50]. This may yield better cosmetic results, since symmetry of the globe position and contour around the orbit is difficult to achieve with bone grafts. Furthermore, malar reduction with osteotomies and orbital wall decompression may be helpful for achieving more symmetry in the malar area with the improvement of exophthalmos [51]. If a lesion is only confined to the maxillary sinus, decortication of the lesion through a window in the anterior wall and replacing the recontoured anterior wall with miniplates can be another simple option [52,53]. Lesions in the naso-ethmoid region can affect the airway and the position of the globe, which can cause significant deformity and functional deterioration. Although diplopia is not a common symptom of craniofacial FD, surgical intervention may be associated with this disabling complication. An otorhinolaryngological evaluation is recommended in addition to a detailed ophthalmological evaluation. Treatment strategies are aimed at: reducing airway obstruction, correcting globe position and visual function, and correcting physical deformities. In this area, surgical options include subtotal excision and radical excision with reconstruction of the skull base and orbits.
In zone 2 (hair-bearing cranium), conservative recontouring procedures are used since this area has fewer cosmetic requirements. The approach is done through coronal incisions, and burring is usually performed to restore a symmetric contour [54]. Complete excision and a bone graft are options from some anterior, more visible areas [39].
In zone 3 (central cranial base, petrous mastoid, and pterygoid bones), surgery should be avoided if possible since there are many important arteries and cranial nerves in this region. Surgery should not be performed in the absence of functional deficits, and observations should be carried out until symptoms occur, at which time decompressive procedures are performed [55]. Prophylactic optic nerve decompression is not recommended even in the setting of radiographic encasement of the optic canal. A meta-analysis including 241 patients with FD suggested that surgery in asymptomatic patients actually increases the risk of vision loss [22,56,57].
The management strategy for optic nerve compression remains a matter of debate. In cases of no evidence of optic neuropathy or subtle and gradual optic neuropathy, routine close observations are warranted, and GH excess should also be checked and managed. Even with complete encasement of the optic nerve by FD, most patients (88% in Cutler and colleagues’ study of 87 patients) have no evidence of optic neuropathy and even when some degree of neuropathy is present, it cannot be definitively explained as resulting from encasement by FD [58]. In cases of acute visual loss or changes, secondary lesions such as an aneurysmal bone cyst and/or mucocele should be evaluated with MRI. Prompt surgical decompression within a week or less is recommended. Medical control of intracanal pressure with acetazolamide is also suggested since the mechanism of acute vision loss is considered to be intralesional hemorrhage of an aneurysmal bone cyst or expansion of the lesion causing acute compression of the optic nerve.
In zone 4 (teeth-bearing bones), a conservative approach is also preferred at the initial stage. Preservation of the original dentition is advisable in any circumstances due to its irrefutably superior function compared to dentures [59]. For the maxilla, by an upper gingivobuccal incision, burring is performed with caution regarding the dental roots and infraorbital nerve. If the buttress is compromised, a bone graft is needed. For the mandible, by a lower gingivobuccal incision, burring is performed with caution regarding dental roots and mental nerve. A symmetrical mandibular contour should be achieved while preserving a stable mandibular arch. If this is not possible, a load-bearing reconstruction plate with a bone graft or flap should be considered. Patients with craniofacial FD have an observed predisposition to malocclusion [19], often requiring orthodontic treatment. Dental procedures are safe to perform in patients with FD and do not adversely affect the course of the disease. Orthognathic surgery, in combination with orthodontic therapy, can also be performed in FD patients at skeletal maturity and disease stabilization [60–62].
Follow-up
To monitor disease progress and the response to nonsurgical treatment, the routine evaluation of serum alkaline phosphatase levels is a reasonable cost-conscious option. Other markers for disease activity are serum osteocalcin, bone-specific alkaline phosphatase, procollagen type 1 N-terminal propeptide, and C-terminal telopeptide. Additionally, urinary hydroxyproline, standard biochemistry screening of the renal profile, serum bicarbonate, albumin-adjusted serum calcium, phosphate, 25OH-vitamin D, GH, and parathyroid hormone levels should be included [20].
CONCLUSION
The craniofacial region is the most commonly involved area in polyostotic FD, and various considerations need to be adopted from other areas. Despite an understanding of the pathophysiology and natural history of FD, there are many unanswered questions about the ideal management method and the expected disease progression. Surgical removal is the standard of care for FD. However, cosmesis is not a sole goal of management. Preservation of function regarding patients’ development through childhood and adolescence depends on collaboration with other specialists. Emerging medical treatments, including immunotherapies and updated technical support for patient-specific surgery, may be promising in achieving more stable disease control and better aesthetic/functional outcomes.
Abbreviations
BMSC
bone marrow stromal cell
cAMP
cyclic adenosine monophosphate
CT
computed tomography
FD
fibrous dysplasia
GH
growth hormone
MAS
McCune-Albright syndrome
MRI
magnetic resonance imaging
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Patient consent
The patients provided written informed consent for the publication and use of their images.
Funding
None.