INTRODUCTION
Angiosarcomas are soft tissue sarcomas that originate from endothelial cells. They are exceedingly rare, accounting for less than 1% of all sarcomas [1]. These tumors can develop anywhere in the body and can occur at any age. However, they most commonly present as cutaneous disease in elderly white men, particularly affecting the head and neck region, especially the scalp. Angiosarcomas can also originate from the breast, soft tissues, bone, and visceral organs [2,3]. Several risk factors for angiosarcomas have been identified, including therapeutic radiation, chronic lymphedema, exposure to various carcinogens, and several genetic syndromes [4,5]. Treatment options include surgical resection for localized disease and systemic chemotherapy for metastatic disease. Achieving negative margins can be challenging due to the infiltrative nature of these tumors. A multidisciplinary approach, which may include surgical resection, chemotherapy, radiotherapy, or potential immune-agents, could yield positive outcomes [4-6]. In this article, we aim to review the fundamental characteristics of angiosarcomas and provide a brief overview of the up-to-date treatment options for this disease.
EPIDEMIOLOGY
Angiosarcomas account for up to 2% of all soft tissue sarcomas and 5.4% of cutaneous soft tissue sarcomas [7,8]. These tumors are more prevalent among the elderly, with the average age of patients being 73, and they exhibit a similar incidence rate in both sexes [9]. However, angiosarcomas of the head and scalp are more common in males, while those associated with radiation and lymphedema are more frequently observed in females [10,11]. Despite the endothelial cell origin of these tumors, it is rare for them to arise directly from major blood vessels or the heart. Instead, they can develop in any soft tissue structure or viscera [3,12].
ETIOLOGY
Angiosarcomas can be categorized into five types: cutaneous angiosarcoma, lymphedema-associated angiosarcoma, radiation-induced angiosarcoma, primary-breast angiosarcoma, and soft tissue angiosarcoma [13]. Several risk factors have been well described in the literature (Table 1). Chronic lymphedema is a well-recognized factor in the development of breast angiosarcomas following breast cancer treatment. Similar conditions, such as Milroy disease, chronic infections, and filariasis, have also been associated with the development of angiosarcomas [4,14]. Likewise, radiotherapy is acknowledged as an independent risk factor for angiosarcomas, although the connection between radiotherapy and subsequent angiosarcoma is disputed, with some suggesting that the risk may stem from concurrent lymphedema [10,11,15-17]. Various familial syndromes, including neurofibromatosis, Maffucci syndrome, and Klippel-Trenaunay syndrome, have been linked to angiosarcoma. Additionally, some case reports have identified mutations in BRCA1 and BRCA2 as potential risk factors for angiosarcomas following breast cancer treatment [18]. Carcinogens such as vinyl chloride, thorotrast, androgenic steroids, phenylhydrazine, arsenic, and radium are considered potential etiological agents in visceral angiosarcoma, possibly accounting for up to 25% of cases [19,20]. Other proposed risk factors include ultraviolet radiation, immunocompromised states, arteriovenous fistulas, and xeroderma pigmentosum [21-23].
HISTOPATHOLOGY
Histologically, the characteristics of angiosarcomas can vary both within a single case and between different cases. It can be challenging to distinguish the morphological features of a malignant vascular tumor from a benign lesion that exhibits proliferation. However, angiosarcomas typically present with abnormal, pleomorphic, malignant endothelial cells. These cells can take on various shapes, including rounded, polygonal, or fusiform, and often exhibit an epithelioid appearance. In areas where the tumor is well-differentiated, it may mimic hemangiomas or lymphangiomas. Here, atypical epithelial cells or diffuse epithelioid and spindle cell proliferations form functioning vascular sinusoids. As the disease becomes more aggressive, these structures grow more complex, with multilayered endothelial cell lining and the formation of papillary projections. Highgrade tumors often exhibit mitotic activity. Indicators of a poorly differentiated, high-grade tumor include necrosis and hemorrhage, along with continuous sheets of endothelial cells [4,5,23,24].
An immunochemical analysis reveals that angiosarcoma typically expresses endothelial markers such as CD31, CD34, von Willebrand factor, Ulex europaeus agglutinin 1, and vascular endothelial growth factor (VEGF). Of these markers, von Willebrand factor, Ulex europaeus agglutinin 1, and CD31 are particularly useful in distinguishing poorly differentiated diseases. Additionally, the absence of melanocytic markers, including S100, human melanoma black-45, and melanoma antigen, can aid in differentiating angiosarcoma from melanoma [25].
MOLECULAR BIOLOGY
While no specific chromosomal abnormalities have been identified in the development of angiosarcoma, certain somatic gene mutations have been reported. Behjati et al. [26] examined 39 angiosarcoma cases and discovered somatic mutations in PTLR and PLCG1, as well as in KRAS, NRAS, HRAS, PIK3CA, and FTL4. Other research has identified mutations in TP53 and CDKN2A, which were the most frequently altered genes in the study, with half of the cases exhibiting MAPK pathway derangements [27]. In addition, several studies have shown that the amplification of the proto-oncogene MYC is commonly found in radiation-induced angiosarcoma, but not in sporadic types [15,27,28]. This broad spectrum of genetic abnormalities may account for the clinical variability of tumor subtypes. As such, the use of gene-expression microarray technology is crucial for identifying molecular markers, and a multidisciplinary approach is necessary for treatment.
CLINICAL PRESENTATION
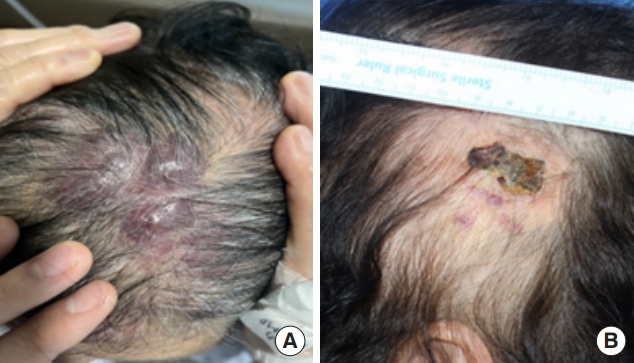
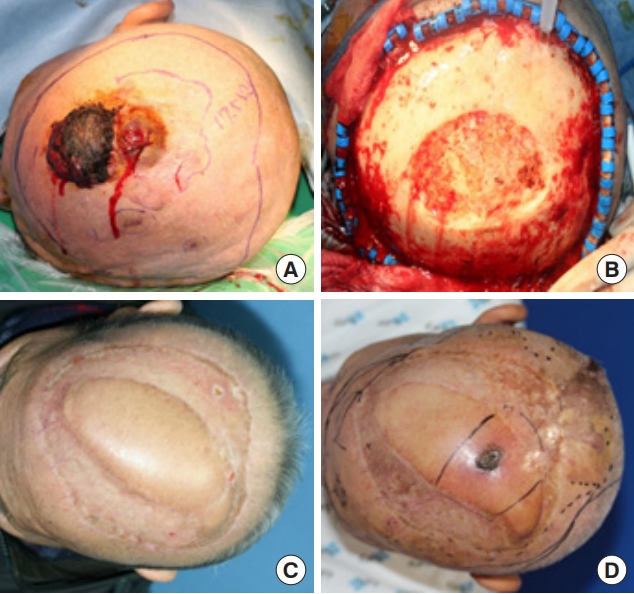
Despite their aggressive nature, angiosarcomas initially present as a bruise, erythematous patch, plaque, or small papule (Fig. 1) [10,24]. Their innocuous clinical appearance often leads to delayed presentation and diagnosis [29]. As the tumor size increases, symptoms such as ulceration, hemorrhage, tissue infiltration, edema, and tumor fungation may occur. The invasion of surrounding structures can result in unclear resection margins, leading to large soft tissue defects following radical resection surgery (Fig. 2) [12,29,30]. The visceral type of angiosarcoma can occur in the spleen, liver, or even the heart. It often presents as an expanding mass associated with pain and discomfort, and rarely with pericardial effusion. Angiosarcomas may also present as pleural disease, hemorrhagic pleural effusion, or pneumothorax due to hematogenous spreading. Other common sites of metastasis include the liver, bone, skin, and regional lymph nodes [3,29,31-33].
PROGNOSIS
Angiosarcomas have a 5-year survival rate that varies between 30% and 56%. Even the most optimistic study indicates that only 60% of patients with localized disease survive beyond 5 years [3,12,31,34]. While there are studies documenting longterm survivors with metastatic disease, they have not been able to identify significant prognostic factors [12]. Numerous studies have sought to uncover the prognostic factors of angiosarcomas. Similar to other aggressive tumor types, age, size, grade, and margin status have been identified as indicators of recurrence and survival [5]. Although angiosarcomas can develop in young patients and even children, it is predominantly a disease of the elderly [35]. A poor prognosis in elderly patients may be due to a longer interval between initial diagnosis and treatment, a more compromised immune system, and a poorer performance status [29]. From the standpoint of tumor biology, a tumor size exceeding 5 cm is associated with worse outcomes [24,36-41]. Tumor depth is another factor linked to a poorer prognosis, but there is no consensus on whether greater tumor depth predicts worse survival or only affects local recurrence [24,31,42]. Other factors that predict a poor prognosis include the metastatic state at presentation and poor patient performance [12,29,43]. Furthermore, the site of origin also influences the prognosis, with angiosarcomas of the liver or other viscera and retroperitoneal disease associated with a worse prognosis compared to cutaneous disease [12].
TREATMENT
While there have been no specific randomized clinical trials for angiosarcomas, treatment has been guided by the published guidelines for other soft tissue sarcomas. There has been no evidence-based management available for patients with this rare disease. However, a multidisciplinary approach to treatment is always recommended to manage the disease and enhance survival outcomes. Currently, more clinical trials are being conducted to broaden treatment options and improve patient outcomes.
Surgery
For localized disease, the standard treatment is radical surgery with a complete resection margin. However, achieving wide negative margins can often be challenging due to tissue infiltration and involvement of anatomical regions such as cardiac tumors and head and neck tumors, which render the disease unresectable [12,29-31]. Choi et al. [44] reported that only a deep margin for excision was significantly related to recurrence. However, the prognostic significance of surgical margins remains to be clarified, and surgical efforts should strive to achieve a microscopically clear margin whenever possible [44,45]. A staged approach may be employed when definitive pathologic margins are determined at the initial resection. In such cases, re-excision can be performed, followed by reconstruction [29].
Several studies have reported that non-metastatic patients who have undergone surgical resection exhibit improved recurrence and survival rates compared to those who have not been resected [28,36,39,46,47]. However, due to the absence of randomized studies, the general approach favors surgical management of angiosarcomas in non-metastatic cases when possible. Nevertheless, other treatment modalities should be considered to enhance patient outcomes [5].
Radiation
Due to the high risk of local recurrence, adjuvant radiation is another method used in the management of angiosarcoma. Although no randomized trials have been conducted, several studies indicate that it enhances local control and overall survival [3,6,29,36,46]. Resection surgery combined with radiotherapy yields better results than either radiation or surgery alone, underscoring the need for a multidisciplinary approach [29,36]. However, in cases of radiation-induced angiosarcoma, radiotherapy is often avoided due to dose-related toxicity. Despite its limited use, radiotherapy should still be considered as an adjuvant measure to reduce the risk of local recurrence in angiosarcomas [5].
Chemotherapy
It is clear that there is no evidence supporting the application of neoadjuvant or adjuvant chemotherapy following resection surgery and radiotherapy for angiosarcoma [32,48-50]. However, systemic chemotherapy is the primary option in advanced and metastatic cases [4-6]. Angiosarcomas have been treated in a manner similar to other soft tissue sarcomas, using an anthracycline-based regimen. The effectiveness of this anthracyclinebased therapy in treating angiosarcoma is comparable to its effectiveness in treating other types of sarcoma. Notably, the combination of ifosfamide with anthracycline has been shown to result in a longer progression-free survival period than treatment with anthracycline alone [51].
The application of pegylated doxorubicin is typically restricted to certain types of soft tissue sarcoma, such as desmoid tumors. Numerous studies have indicated the potential efficacy of pegylated doxorubicin in treating angiosarcomas. Therefore, it could be a viable alternative for patients who are unable to withstand more aggressive chemotherapy treatments [52].
Paclitaxel is frequently employed as a first or second-line treatment for angiosarcoma due to the antiangiogenic activity of taxanes. The effectiveness of paclitaxel was evaluated in a multicenter phase II study involving 30 angiosarcoma patients. The study reported an overall response rate of 19% at 6 months. Several retrospective studies have also confirmed the efficacy of paclitaxel [53-55].
Gemcitabine combined with docetaxel is a common secondline treatment for advanced soft tissue sarcomas, yet no prospective studies exist for metastatic angiosarcoma. Recently, an alternative combination of gemcitabine with nanoparticle albuminâ bound paclitaxel (nab-paclitaxel) was explored. The overall better toxicity profile of nab-paclitaxel compared to paclitaxel may offer an alternative option. Future studies should consider these combinations for use in advanced angiosarcomas [56,57].
Eribulin, an agent that disrupts microtubule polymerization, has recently been reported in cases of cutaneous angiosarcoma that have progressed with taxanes. This agent, which exhibits less neurotoxicity than taxanes, may provide a treatment option for cases that have shown progression despite treatment with taxanes and are not eligible for an anthracycline-based regimen [58].
Encequidar is an adenosine triphosphate binding cassette transporter P-gp inhibitor that prevents the efflux of cytotoxic agents from epithelioid cells to the gastrointestinal tract, thereby promoting increased oral bioavailability and enhancing the efficacy of chemotherapy. A recent phase II study investigated the combination of encequidar and oral paclitaxel for patients with unresectable cutaneous angiosarcoma who had not previously undergone taxane therapy. The study demonstrated favorable side effect profiles, particularly considering the notably advanced median age of the patients [59].
Targeted therapy
In angiosarcomas, VEGF and its receptors are overexpressed, making the targeting of this pathway a feasible option. Bevacizumab, a VEGF inhibitor, has demonstrated a modest effect in advanced angiosarcoma [60]. The combination of bevacizumab with paclitaxel, gemcitabine, or docetaxel has been tested in several studies. However, whether these combination strategies exhibit strong activity requires further large-scale prospective investigation [61,62].
Tyrosine kinase inhibitors (TKIs) possess anti-VEGF activity and have been the subject of studies in angiosarcoma. Pazopanib, a TKI approved for use in soft tissue sarcomas, has demonstrated modest benefits in retrospective studies. An antibody to endoglin, known to mediate resistance to pazopanib, was examined in a phase III trial but did not exhibit significant activity [63-66]. Regorafenib, another TKI with anti-VEGF activity, showed some effectiveness in a small number of angiosarcoma patients [67]. Propranolol, which is typically effective in treating benign vascular tumors, has been investigated and utilized in the treatment of some angiosarcoma patients [68].
Immunotherapy
Pembrolizumab, an anti-PD1 (programmed death 1) checkpoint inhibitor, has been approved for use in tumors with a high tumor mutational burden. It was previously reported that this checkpoint inhibitor was effective in treating three out of ten angiosarcoma patients who showed no response to standard therapies [69,70]. Other checkpoint inhibitors, such as anti-PD-L1 or anti-CTLA-4 agents, have also been reported to elicit varying responses in cutaneous angiosarcomas and radiation-associated breast angiosarcomas [71-73]. Current investigations are focusing on using immunotherapy alone or in combination with systemic chemotherapy, TKIs, or an oncolytic virus to treat angiosarcomas.
Regional therapy
Isolated limb perfusion (ILP) is a procedure that involves administering chemotherapy drugs directly into a limb affected by locoregional metastasis. ILP has been explored as an alternative treatment when radical surgery aimed at preserving function is not viable. While some studies have demonstrated the effectiveness of ILP, its use can be challenging due to the risk of severe complications and a general lack of expertise in the technique [74]. For primary liver angiosarcomas, transarterial embolization has shown minimal impact on survival rates. However, intra-arterial therapies may be a viable option when it is necessary to manage liver disease [75-77].
CONCLUSION
Angiosarcoma is an extremely rare soft tissue sarcoma, characterized by its complex biology and aggressive nature. For the treatment of angiosarcoma without clear metastasis, a wide margin resection is necessary, coupled with a staged multimodal approach. The use of both radiotherapy and systemic chemotherapy is advisable to enhance outcomes, while antiangiogenic biological therapies may provide specific treatments for angiosarcoma. Further clinical trials, grounded in translational work, hold the potential to significantly influence and clarify treatment strategies.